


Kurt de VLAM1, Alice B. GOTTLIEB2 and Philip J. MEASE3,4
1Department of Rheumatology, University Hospitals, Leuven, Belgium, 2Department of Dermatology, Tufts Medical Center, Boston, Massachusetts, 3Division of Rheumatology Research, Swedish Medical Center, and 4Division of Rheumatology, University of Washington School of Medicine, Seattle, Washington, USA
Psoriatic arthritis occurs in a subset of psoriasis patients and is therefore commonly encountered in dermatology practice. Although its exact pathogenesis is unknown, psoriatic arthritis is thought to share common mechanisms with psoriatic skin symptoms. Innate and adaptive immune responses are abnormally activated in psoriasis and may acquire the ability to attack peripheral joints and other sites following an environmental trigger (e.g. mechanical stress, trauma, infection) in genetically susceptible patients. The increased cardiovascular risk inherent in psoriasis appears further enhanced in psoriatic arthritis, likely reflecting the overall burden of systemic inflammation contributing to atherogenic processes. Basic research and clinical trials have suggested that tumour necrosis factor is important in psoriatic arthritis pathophysiology, and accumulating evidence suggests that Th17 cells and interleukin-17A may also be important. Basic research and clinical trials inform our understanding of psoriatic arthritis pathophysiology and, in turn, help dermatologists to make better treatment decisions. Key words: biologics; cardiovascular disease; IL-17A inhibitors; pathophysiology; psoriatic arthritis; TNF inhibitors.
Accepted Feb 26, 2014; Epub ahead of print Feb 27, 2014
Acta Derm Venereol
Kurt de Vlam, MD, PhD, Department of Rheumatology, University Hospitals Leuven, Herestraat 49, BE-3000 Leuven, Belgium. E-mail: Kurt.devlam@uzleuven.be
Psoriatic arthritis (PsA) is a form of chronic arthritis associated with psoriasis (1). Cutaneous manifestations are seen well before the appearance of joint symptoms in most cases, although approximately 14–20% of patients present with PsA before or simultaneously with skin symptoms (2). When psoriatic skin symptoms occur first, the mean time to PsA appearance is 10–12 years (3, 4). Clinically, PsA is characterized by peripheral and/or axial joint inflammation with associated pain and/or tenderness (Fig. 1) (1, 5, 6). All 3 joints of a digit may be affected while adjacent digits are spared. Radiographically, the hallmark of PsA is the simultaneous presence of bone resorption and bony proliferation (1). Other common features include dactylitis (i.e. inflammation of an entire digit, creating a sausage-like appearance) and enthesitis (i.e. inflammation at attachment sites for tendons, ligaments, or joint capsule fibers); each may occur in up to 50% of PsA patients (7). Spondylitis (i.e. inflammation of the spine and the sacroiliac joint) may also be present in PsA but is typically less prominent than peripheral joint disease (7). The clinical course of PsA is highly variable. Some patients have a mild, nondestructive clinical phenotype, but others develop progressive joint damage that if left untreated can lead to functional impairment and disability (8).
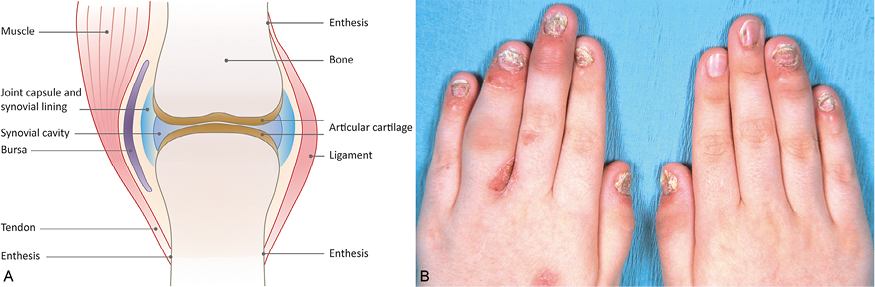
Fig. 1. Schematic of joint structure (A) and photograph of common symptoms in psoriatic arthritis (B). The symptoms shown include erythema and swelling of the distal interphalangeal joints in both hands, diffuse swelling of the left 4th proximal interphalangeal joint producing a sausage-like appearance (dactylitis), and nail changes typical of psoriasis. B: ©2013 American College of Rheumatology. Used with permission.
In this article, we provide an overview of PsA pathophysiology, drawing connections between disease mechanisms in the skin and joints and discussing the relationships of these mechanisms with premature cardiovascular disease. We then describe the results of recent clinical trials, focusing on interleukin (IL)-12/IL-23 inhibitors and IL-17A inhibitors, which may shed additional light on PsA pathophysiology.
PATHOGENESIS OF PSORIATIC ARTHRITIS
Risk factors
Patients with greater psoriatic skin involvement are at increased risk of developing PsA, as illustrated by results from a U.S. population-based study (Fig. 2) (9). Similarly, in European dermatology clinic patients, greater body surface area (BSA) involvement, both at the time of the study assessment and within 6 months after psoriasis diagnosis, was associated with PsA risk (10). However, the extent and severity of psoriasis quantified by the Psoriasis Area and Severity Index (PASI) do not correlate with the severity of PsA joint disease (11). Specific psoriasis features independently associated with PsA risk include scalp lesions (by 3.75-fold), nail dystrophy (by 2.24-fold), and intergluteal/perianal lesions (by 1.95-fold) (12). From an anatomical perspective, the nail is linked to the entheses via fibers from the extension tendon and the collateral ligament of the distal interphalangeal joint. It has been proposed that tissue-specific factors, including microtrauma and biomechanical stress, cause aberrant innate immune responses and persistent inflammation, thereby potentially linking the nail changes in psoriasis with enthesitis and joint involvement in PsA (13, 14).
Epidemiologic studies have shown a positive correlation between body mass index and the risk of incident PsA in psoriasis populations (15, 16). In addition to being a risk factor for PsA, obesity may also adversely impact the efficacy of tumour necrosis factor (TNF) inhibitors in decreasing disease activity in PsA, although the evidence is inconclusive (17, 18). A potential mechanism for these hypothesized effects of obesity is the release of adipokines (including TNF) by fat cells (19).
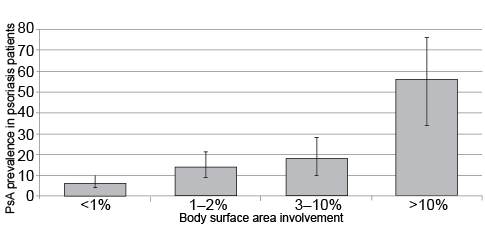
Fig. 2. Prevalence of psoriatic arthritis in psoriasis patients based on the degree of skin involvement. Vertical lines show 95% confidence interval of the prevalence estimate (9).
Genetic factors
Both psoriasis and PsA show high rates of familial aggregation, suggesting that both disorders have a strong genetic basis (20–25). In a landmark study by Moll & Wright (20), the prevalence of PsA among first-degree relatives was 49 times higher than the prevalence in the general population. Genome-wide association studies (GWAS) indicate that specific class I human leukocyte antigen (HLA) genes in the major histocompatibility complex (MHC) are highly associated with PsA and may account for approximately 30% of the genetic susceptibility (26, 27). Two different patterns of HLA involvement have emerged. HLA-Cw*0602, the major determinant of susceptibility to psoriatic disease, confers susceptibility to a phenotype with more severe skin symptoms and musculoskeletal involvement appearing 10 or more years after the onset of psoriasis (27). In contrast, certain HLA-B alleles (e.g. HLA-B*27, B*38, B*08) may be associated with more prominent musculoskeletal features that occur closer to the time of psoriasis onset. A family-based association study showed that the HLA-B*27, B*38, B*39, and C*12 alleles are significantly over-transmitted in PsA compared with psoriasis (28). Moreover, certain HLA-B and HLA-C haplotypes are highly associated with PsA susceptibility among psoriasis patients (29). For example, the odds ratios for PsA were 41 and 20 when HLA-B*27 was paired with HLA-C*01 or HLA-C*02, respectively.
GWAS have also identified other susceptibility genes for psoriasis and PsA, which can be grouped into an integrated pathogenic disease model comprising distinct signalling networks that affect skin barrier function, innate immune responses involving nuclear factor κB (NF-κB) and interferon (IFN) signalling, and adaptive immune responses involving CD8+ T cells and CD4+ T helper (Th)17-cell signalling (Fig. 3) (30). Many of these mechanisms had been identified years before the GWAS. For example, TNF activates multiple signalling pathways in psoriasis and PsA, including NF-κB. Whereas NF-κB was not activated in epidermis from normal individuals, increased numbers of keratinocytes containing activated NF-κB were detected in uninvolved epidermis of psoriasis patients and even higher upregulated levels were found in the epidermis of psoriatic plaques (31). Similarly, activated NF-κB was found in the synovial lining and in infiltrating and perivascular cells in the sub-lining zone of PsA patients (32). In both cases, a TNF inhibitor significantly reduced NF-κB signalling (31, 32).
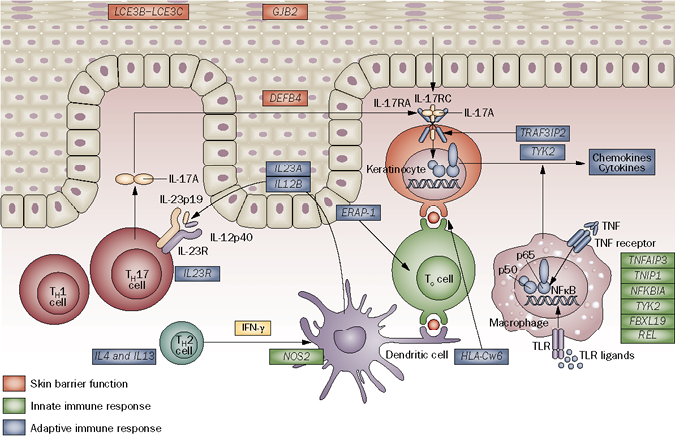
Fig. 3. Susceptibility genes for psoriasis, and by analogy for psoriatic arthritis, grouped into an integrated pathogenic disease model comprising distinct signalling networks that affect skin barrier function, innate immune responses and adaptive immune responses. Genes in which variants have been associated with psoriasis are color-coded according to the signalling pathway in which they function. IFN: interferon; IL-17: interleukin-17; NFκB: nuclear factor κB; Tc cell: cytotoxic T cell; TH1 cell: T helper 1 cell; TH2 cell: T helper 2 cell; TH17 cell: T helper 17 cell; TLR: Toll-like receptor; TNF: tumour necrosis factor. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Rheumatology 7: 718–732, copyright 2011.
Most susceptibility loci for PsA show overlap for psoriasis, consistent with the clinical and genetic relationships between the two diseases. However, as noted above, HLA-B*27 and other HLA-B alleles have been strongly associated with PsA and not with psoriasis. In addition, polymorphisms in the IL-13 gene (which encodes a Th2 cytokine) have been associated with PsA and not psoriasis, specifically among non-smokers (33–35).
Environmental factors
Environmental factors appear to play a role in PsA pathogenesis in individuals with appropriate genetic susceptibility to psoriatic disease. The identification of a viral replication marker in blood samples of PsA patients but not of rheumatoid arthritis (RA) patients led to the suggestion that viruses may affect the risk for PsA (36), although there have been no confirmatory data since this finding. Two other observations support potential viral involvement. First, human immunodeficiency virus (HIV) exacerbates psoriasis and PsA (37, 38), and second, a balance between activating and inhibitory killer-cell immunoglobulin (Ig)-like receptors (KIRs) on natural killer (NK) cells combined with specific HLA class I molecules appears to be important in conferring PsA susceptibility (39, 40). Nonetheless, no data exist to indicate an epidemiological association between viral infection and psoriasis or PsA.
Evidence for the role of trauma is more convincing. Case series suggest that recent trauma may precede the onset of PsA in 8–9% of patients (41, 42). In case-control studies, several factors have been independently associated with recent-onset PsA in psoriasis patients, including injury, rubella vaccination, recurrent oral ulcers, moving house, lifting heavy cumulative loads, and infections requiring antibiotics (43, 44). The Koebner phenomenon, in which trauma to non-lesional skin can induce a plaque in psoriasis patients, is well established. It is possible that trauma to the joint may cause a similar phenomenon.
Cardiovascular disease
PsA is associated with increased cardiovascular morbidity, including coronary heart disease, congestive heart failure, cerebrovascular disease, and peripheral vascular disease (45–47). Higher cardiovascular risk in PsA reflects an increased prevalence of cardiovascular risk factors and surrogate markers of subclinical atherosclerosis compared with control populations. Traditional cardiovascular risk factors more common in PsA than in the general population include hypertension, dyslipidemia, diabetes, glucose intolerance, obesity, and the metabolic syndrome (45, 46, 48–50). Surrogate markers of subclinical atherosclerosis associated with PsA include increased carotid intima-media thickness (48), endothelial dysfunction (51), and increased ventricular-arterial stiffness (52).
C-reactive protein (CRP), a marker of systemic inflammation, is well recognized as a nontraditional cardiovascular risk factor (53). CRP and other systemic inflammation markers are higher in PsA patients than in healthy subjects (48) and psoriasis patients (54, 55). In an experimental model, chronic skin inflammation promoted vascular inflammation and thrombosis and, conversely, treatment of skin inflammation reduced the vascular changes (56). Similarly, CRP levels in PsA and psoriasis patients were reduced following TNF inhibitor therapy (54).
A concept termed the “psoriatic march” has been proposed to causally link PsA (as well as psoriasis) with increased cardiovascular risk. According to this model, psoriatic disease produces systemic inflammation, which causes insulin resistance and endothelial dysfunction, which lead to atherogenesis and ultimately to major cardiovascular events (57). Retrospective cohort studies suggest that TNF inhibitors may reduce the cardiovascular burden associated with psoriasis (58–60), whereas data from prospective registries of RA patients treated with TNF inhibitors are conflicting and inconclusive (61–63). Cardiovascular benefit with TNF inhibitors remains to be demonstrated in PsA.
Cellular mechanisms
Abnormal activation of the innate and adaptive immune systems contributes to chronic disease processes in both psoriasis and PsA (64). An environmental triggering event produces damage and this damage initiates an innate immune response in genetically susceptible individuals. This process is better defined in the skin, although parallel mechanisms may operate in the joint. Damage to keratinocytes elaborates antibacterial peptides that activate skin plasmacytoid dendritic cells to produce IFN-α; it also stimulates other innate immune cells in the skin to produce additional pro-inflammatory cytokines. In turn, these cytokines stimulate dermal myeloid dendritic cells to migrate to skin-draining lymph nodes, where they present unknown antigens to drive T-cell activation and proliferation. Subsequently, the expanded T-cell populations return to the skin to perpetuate and amplify inflammatory mechanisms.
In PsA, the skin and the joints exhibit a prominent lymphocytic infiltrate consisting of activated CD4+ and CD8+ T cells as well as an increase in neutrophil infiltration (1, 65). CD4+ cells predominate in the dermal papillae of the skin and the sub-lining stroma of the joint, whereas CD8+ cell infiltration is prominent in the epidermis and the inflamed enthesis. Neutrophils are localized to epidermal micro-abscesses in psoriatic skin and to synovial tissue in the joint (66, 67).
CD4+ differentiation was initially classified according to a Th1–Th2 paradigm (68), but a third T-helper subset was identified in 2005, whose characteristic product is IL-17A (69, 70). These Th17 cells are increased in the circulation of psoriasis and PsA patients and are also increased in the synovial fluid of PsA patients compared with osteoarthritis patients (71–73). Other IL-17A-positive cells, including neutrophils and/or mast cells, have been identified in psoriatic skin lesions and PsA synovium (66, 74). IL-17A expression in skin lesions correlates with disease activity in psoriasis (65). The role of this cytokine in the pathogenesis of psoriasis has been characterized in considerable detail. IL-17A acts on a variety of skin cell types, including endothelial cells, fibroblasts, monocytes, and keratinocytes, stimulating the production of pro-inflammatory antimicrobial peptides, chemokines and angiogenic factors, which in turn promote the recruitment of inflammatory cells and establish a self-sustaining feedback loop (75). Other cytokines structurally related to IL-17A have been identified (IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F), although their functional roles have been less well characterized. IL-17B, IL-17C, and IL-17F are known to induce the expression of inflammatory cytokines and promote neutrophil infiltration and so may also contribute to psoriasis pathogenesis (76). IL-17F is highly homologous to IL-17A and can exist as a heterodimer with IL-17A (77). IL-17F is less potent than IL-17A in inducing inflammatory cytokine expression, with the IL-17F/IL-17A heterodimer exhibiting intermediate potency (78).
The PsA synovium, like psoriatic skin, is characterized by the over-expression of multiple cytokines, including TNF, IFN-γ, IL-1β, IL-6, IL-10, IL-15, and IL-18 (79, 80). Many of these cytokines have pleiotropic effects, which include the upregulation of chemokines and cell adhesion molecules to help guide cell infiltration to the site of inflammation. IL-23, a key cytokine promoting the Th17 phenotype, is significantly increased in psoriasis lesions compared with non-lesional and normal skin (81, 82), and increased synovial expression of the p19 subunit of IL-23 is associated with markers of PsA activity (83). In an animal model of spondyloarthritis (SpA), IL-23 acted on entheseal resident T cells to promote pathogenic changes characteristic of the human disease, with accompanying production of several cytokines, including IL-17A (84).
Cytokines such as TNF and IL-1β increase the production of matrix metalloproteinases (MMPs) from macrophages (5). Several MMPs are elevated in PsA and contribute to cartilage degradation (85, 86). TNF also increases the expression of vascular endothelial growth factor and other vascular markers, which are seen in both psoriatic skin and PsA synovium and may contribute to the increased vascularity at both locations (79, 87). Moreover, TNF promotes osteoclast precursor differentiation and migration to inflamed synovium and subchondral bone, resulting in progressive bone damage (88). IL-17A also acts on multiple cell types, including keratinocytes in the skin and synovial-like fibroblasts in the joint, to increase the production of mediators that help sustain the chronic inflammatory state (89–91). In addition, IL-17A acts on osteoblasts and osteoclast precursors to promote bone resorption, which may contribute to joint damage in PsA (92, 93). Data from an animal model of SpA indicate that IL-22 may mediate bone formation (84).
Interventional clinical studies provide evidence for the importance of these cellular mechanisms in psoriasis and PsA. TNF inhibitors reduce T-cell infiltrate in the skin and synovium as well as synovial macrophage infiltrate (94, 95). Moreover, they slow or prevent radiographic disease progression in PsA (96–98). Specifically, TNF inhibitors decrease bone erosion, but it is unclear whether they also ameliorate bony spur formation along the entheses. Finally, in a small study of 9 PsA patients, TNF inhibitors significantly reduced the mitogen-activated protein kinases (MAPK) ERK and JNK, but not p38, in PsA synovium. Persistent activation of intracellular signalling pathways, even when reduced by TNF inhibitors, may potentially explain the short time to relapse after stopping treatment (32).
OVERVIEW OF PSORIATIC ARTHRITIS MANAGEMENT
Dermatologists should be alert to joint symptoms in patients with psoriasis, especially: 1) stiffness that lasts more than 30–45 min in the morning or after periods of inactivity (e.g. air or car travel); and 2) joint swelling (Table I). Symptoms useful in the diagnosis of PsA include dactylitis and enthesitis, which are uncommon in RA and generally not present in osteoarthritis. The presence of nail lesions can also help distinguish PsA from other forms of coincident arthritis in psoriasis patients. Involved joints in PsA are often less symmetrical in distribution than in RA, although a minority of PsA patients, especially female patients, have a symmetrical polyarticular inflammatory arthritis resembling RA. Signs specific to RA include the presence of rheumatoid nodules and high titers of rheumatoid factor (1). Regarding the assessment of PsA severity, treatment guidelines designate involvement of fewer than 5 joints and response to non-steroidal anti-inflammatory drug (NSAID) therapy as indicative of “mild” PsA, whereas 5 or more swollen or tender joints, inadequate response to NSAIDs, and the presence of joint damage and functional impairment are signs of more severe PsA (1, 99, 100).
Table I. Symptoms useful for psoriatic arthritis screening in the dermatology clinic (1)
|
Joint stiffness |
Lasting more than 30–45 min Occurring in the morning or after periods of inactivity |
|
Joint swelling |
Dactylitis Enthesitis (especially at the insertions of the Achilles tendon and the plantar fascia) |
|
Nail lesions |
Pitting Discoloration Thickening Deformation Onycholysis |
The diagnosis of PsA nevertheless remains challenging, as the disease has a heterogeneous clinical phenotype with respect to joint involvement. Five clinical patterns of PsA were originally described by Moll & Wright (101), who considered asymmetric oligoarthritis to be the most common presentation. Later research suggested that a symmetric polyarthritis is more common (102). In fact, clinical phenotypes can overlap and change over time in a single patient. Oligoarthritis may, for example, appear early in the disease and eventually progress to a polyarticular pattern as more joints are affected. Consequently, it has been suggested that there may be little clinical utility in detailed phenotypic classification systems, and that distinguishing between PsA affecting peripheral joints only and PsA with axial involvement is sufficient (103). Controversy over PsA classification also exists in relation to the place of PsA within the larger SpA group of diseases, which includes ankylosing spondylitis, reactive arthritis, and SpA related to inflammatory bowel disease. Given the clinical and pathophysiological similarities among PsA and the other SpA, there is uncertainty over whether PsA should be conceptualized as a discrete disease within the SpA category or, by contrast, whether all SpA can be considered manifestations of a single disease (103).
The development of the ClASsification criteria for Psoriatic ARthritis (CASPAR) criteria has facilitated the diagnosis of PsA. The CASPAR criteria have been shown to be highly effective in distinguishing patients with PsA from those with other inflammatory arthritides (104). In a simple scoring system, point values are assigned based on 5 characteristics, including personal or family history of psoriasis, psoriatic nail dystrophy, negative rheumatoid factor test, history of dactylitis, and radiographic evidence of juxtaarticular new bone formation.
PsA management consists of NSAIDs for mild joint symptoms, conventional disease-modifying anti-rheumatic drugs (DMARDs) for moderate-to-severe disease, and TNF inhibitors following the failure of one or more DMARDs (Table II) (1, 99, 100). However, TNF inhibitors can be considered as initial DMARD therapy for patients with very active disease and those with significant enthesitis, dactylitis, or axial disease. It should also be noted that the long-term use of NSAIDs is associated with an increased risk of cardiovascular events (105) and so may not be appropriate for some patients with PsA, which is itself associated with an elevated cardiovascular burden. Traditional oral DMARDs such as methotrexate, sulfasalazine, and leflunomide are used widely as first-line systemic therapy for PsA and remain a mainstay of therapy in locations with limited access to biologics. However, clinical trial evidence of their efficacy is scant and in the case of methotrexate inconclusive (6, 106). Methotrexate has never been shown prospectively to inhibit structural damage in PsA (6).
Table II. Treatment recommendations for PsA (1, 99, 100)
|
Drug class |
AAD (2008) (1) |
GRAPPA (2009) (99) |
EULAR (2012) (100) |
|
NSAIDs |
For relief of mild PsA symptoms |
For mild joint symptoms but not skin symptoms |
For relief of musculoskeletal signs and symptoms |
|
Conventional DMARDs |
For moderate-to-severe PsA that is inadequately controlled by NSAIDs or local steroid injections |
For moderate-to-severe peripheral arthritis; for mild arthritis that does not respond to NSAIDs or local steroid injections |
For patients with active disease, particularly many swollen joints, structural damage with inflammation, high ESR/CRP or clinically relevant extra-articular manifestations |
|
TNF inhibitors |
For moderate-to-severe PsA, usually in combination with DMARD therapy No specific treatment recommendations made based on presence of enthesitis, dactylitis or |
For moderate-to-severe peripheral arthritis that does not respond to ≥ 1 DMARD For DMARD-naïve patients with poor prognosis For patients with severe enthesitis or moderate-to-severe axial disease, which does not respond to NSAIDs or physical therapy For patients with dactylitis that does not respond to NSAIDs or local steroid injections |
For patients with active arthritis who have inadequate response to ≥ 1 DMARD For DMARD-naïve patients with very active disease in exceptional cases For patients with active enthesitis and/or dactylitis and inadequate response to NSAIDs or local steroid injections For patients with predominantly axial disease and inadequate response to NSAIDs |
|
IL-12/IL-23 inhibitor (ustekinumab) |
Ustekinumab was recently approved as a therapy for PsA and accordingly is not included in the treatment recommendations cited here. Appropriate use of ustekinumab will be addressed in updated treatment recommendations. |
||
AAD: American Academy of Dermatology; DMARD: disease-modifying antirheumatic drug; ESR-CRP: erythrocyte sedimentation rate/C-reactive protein; EULAR: European League Against Rheumatism; GRAPPA: Group for Research and Assessment of Psoriasis and Psoriatic Arthritis; IL: interleukin; NSAIDs: nonsteroidal anti-inflammatory drugs; PsA: psoriatic arthritis; TNF: tumour necrosis factor.
TNF inhibitors are active against all disease characteristics in psoriatic disease, including synovitis, joint damage, skin symptoms, nail changes, enthesitis, dactylitis and axial disease (99, 100). However, not all patients achieve clinically meaningful responses, and some may respond initially but lose treatment responses over time. Switching to a second TNF inhibitor is one option (107, 108), or use of an alternative biologic can be considered (109). Finally, some patients have medical contraindications to TNF inhibitor therapy (e.g. congestive heart failure, multiple sclerosis). This underscores the need for new, targeted approaches with proven efficacy and safety in PsA. In turn, proof of the efficacy of such agents should help further our understanding of PsA pathophysiology.
TH17–IL-17A PATHWAY AS A PATHOGENIC TARGET
Several clinical trials in PsA with agents targeting the Th17–IL-17A pathway have been completed and others are in progress. Ustekinumab is an anti–IL-12/IL-23 monoclonal antibody that targets the p40 subunit common to both cytokines and thereby blocks Th1 and Th17 differentiation (110). This agent was originally approved for use in moderate-to-severe plaque psoriasis, where it has efficacy comparable to the anti-TNF monoclonal antibodies. Phase III clinical trials of ustekinumab in PsA were subsequently conducted and also demonstrated efficacy (109, 111). In PSUMMIT 1, ustekinumab significantly increased American College of Rheumatology (ACR) and Disease Activity Score (DAS) 28–CRP response rates and produced greater median changes in the Health Assessment Questionnaire (HAQ) compared with placebo. Comparable results were found in PSUMMIT 2, which extended to the patient subset previously treated with TNF inhibitors. Ustekinumab was also more effective than placebo against skin disease, enthesitis, and dactylitis in patients with these PsA features. Consequently, ustekinumab was approved in September 2013 by regulatory authorities in North America and the European Union as a treatment for PsA (112).
Several IL-17A pathway inhibitors have exhibited promising activity in moderate-to-severe psoriasis in phase II trials, including secukinumab (113, 114), ixekizumab (115), and brodalumab (116) (Table III). Secukinumab and ixekizumab target IL-17A, whereas brodalumab targets a receptor subunit through which IL-17A and other IL-17 cytokines exert their biological effects. Secukinumab and brodalumab are under evaluation in PsA (phase III and phase II trials, respectively). No IL-17A inhibitors are as yet approved for the treatment of psoriasis or PsA.
Table III. Effects of interleukin (IL)-17A inhibitors in randomized controlled phase II trials in patients with moderate-to-severe psoriasisa
|
Drug |
Regimen |
Patients (n) |
Primary endpoint analysis |
Most common adverse eventsb |
|
Secukinumab (113) |
25 mg at week 0, or 25, 75 |
125 |
PASI 75: 57% for 75 mg and 82% for 150 |
Nasopharyngitis (13% vs. 9%), upper respiratory tract infection (8% vs. 0%) |
|
Secukinumab (114) |
150 mg at week 0, or at weeks 0, 4 and 8, or at weeks 0, 1, 2 and 4 |
404 |
PASI 75: 11%, 42% and 54% vs. 2% for |
Nasopharyngitis (20% vs. 18%), headache (7% vs. 4%), upper respiratory tract infection (3% vs. 9%) |
|
Ixekizumab (115) |
10, 25, 75 or 150 mg at weeks |
142 |
PASI 75: 29%, 77%, 83% and 82% vs. 8% |
Infection or infestation (33% vs. 26%), headache (9% vs. 4%) |
|
Brodalumab (116) |
70, 140 or 210 mg at weeks 0, |
198 |
PASI change from baselinec: 45%, 86%, |
Nasopharyngitis (8% vs. 8%), upper respiratory tract infection (8% vs. 5%) |
aPrevious psoriasis medications were first discontinued, and then each agent or placebo was administered subcutaneously. The primary endpoint analyses were conducted at week 12.
bData shown for IL-17A inhibitor groups combined vs. placebo.
cPsoriasis Area and Severity Index (PASI) 75 was evaluated as a secondary endpoint; values for comparison were 33%, 77%, 82%, and 67% vs. 0% for placebo (p < 0.001 for each comparison vs. placebo).
Data on the preliminary safety and efficacy of secukinumab 10 mg/kg in a randomized controlled PsA trial were recently published (117). The ACR20 response rate favoured secukinumab over placebo at week 6, week 12, and week 24, although statistical significance was not achieved at the week-6 primary endpoint, likely reflecting the small sample size. CRP and erythrocyte sedimentation rate (ESR) were decreased in the secukinumab group but not in the placebo group (117).
On the basis of the role of Th cells in host defense, safety concerns with ustekinumab and the IL-17A inhibitors are likely to be similar to those for the TNF inhibitors, namely, serious infection and possibly malignancy. The accurate evaluation of these risks will require long-term exposure in large numbers of patients in real-world clinical practice.
Several small molecules that may indirectly influence the Th17–IL-17A pathway are also under evaluation in PsA (e.g. the phosphodiesterase 4 inhibitor apremilast) or will likely be evaluated on the basis of promising phase II data in psoriasis (e.g. the Janus kinase inhibitor tofacitinib). Phase III data presented in late 2012 showed that apremilast 20 or 30 mg b.i.d. significantly improved ACR response rates and DAS28 and Health Assessment Questionnaire-Disability Index (HAQ-DI) scores compared with placebo (118).
CONCLUSIONS
Basic research and clinical trials provide a two-way approach to a better understanding of disease pathophysiology. Basic research identifies possible mechanisms in disease pathophysiology, and interventional clinical trials can determine whether those mechanisms are important and, if so, for which disease features and in what proportion of patients. Along these lines, the efficacy of TNF inhibitors suggests that TNF plays a role in disease pathophysiology, at least in some PsA patients. Ongoing clinical trials will show whether Th17 cells and IL-17A can also provide the basis for effective therapeutic intervention.
ACKNOWLEDGEMENTS
The authors would like to thank Barry Weichman for assistance in drafting the manuscript and Andrew Horgan of BioScience Communications, Inc, for editorial support.
Funding sources: This manuscript was supported by a grant from Novartis. No compensation was paid to the authors.
Conflicts of interest: Dr. de Vlam has been a consultant, investigator, speaker and/or advisory board member for Pfizer, Abbott, Janssen, MSD, and Novartis and has received compensation in the form of grants and/or honoraria from these companies. Dr. Gottlieb is a consultant and/or advisory board member for Abbott Laboratories, AbbVie, Actelion, Akros, Amgen, Astellas, Beiersdorf, Bristol-Myers Squibb, Can-Fite, Celgene, Coronado, CSL Behring Biotherapies for Life, DermiPsor, GlaxoSmithKline, Incyte, Janssen, Karyopharm, Lilly, Novartis, Novo Nordisk, Pfizer, Teva, UCB, Vertex, and Xenoport, and has received research/educational grants (paid to Tufts Medical Center) from Abbott Laboratories, Amgen, Celgene, Coronado, Janssen, Levia, Lilly, Novartis, and Pfizer. Dr. Mease has been a consultant, investigator, speaker and/or advisory board member for Abbvie, Amgen, Biogen Idec, Bristol-Myers Squibb, Celgene, Genentech, Janssen, Lilly, Merck, Novartis, Pfizer, and UCB and has received compensation in the form of grants and/or honoraria from these companies.
REFERENCES

