


Sigurd Broesby-Olsen1, Ingunn Dybedal2, Theo Gülen3, Thomas K. Kristensen4, Michael B. Møller4,
Leena Ackermann5, Maria Sääf6, Maria Karlsson7, Lone Agertoft8, Kim Brixen9, Pernille Hermann9, Eva
Stylianou10, Charlotte G. Mortz1, Trine Torfing11, Troels Havelund12, Birgitta Sander13, Anna Bergström14, Marie
Bendix15, Lene H. Garvey16, Ole Weis Bjerrum17, Peter Valent18, Carsten Bindslev-Jensen1, Gunnar Nilsson19, Hanne
Vestergaard20 and Hans Hägglund21
Departments of 1Dermatology and Allergy Centre and 4Pathology, 8Hans Christian Andersen Children’s Hospital, Departments of 9Endocrinology, 11Radiology, 12Gastroenterology and 20Haematology, Odense University Hospital, Odense, Denmark, 2Department of Hematology, Oslo University Hospital, Rikshospitalet, Oslo, Norway, Departments of 3Respiratory Medicine and Allergy, 6Endocrinology, Metabolism, and Diabetes, 7Dermatology and Venereology and 15Consultation Psychiatry, Psychiatry Southwest, Karolinska University Hospital, Stockholm, Sweden, 5Department of Dermatology, Skin and Allergy Hospital, Helsinki University Central Hospital, Helsinki, Finland, 10Department of Pulmonary Diseases, Regional Unit for Asthma, Allergy and Hypersensitivity, Oslo University Hospital, Ullevål, Oslo, Norway, 13Department of Laboratory Medicine, Division of Pathology and 19Clinical Immunology and Allergy, Department of Medicine, Karolinska Institutet and Karolinska University Hospital, Stockholm, 14Department of Dermatology and Venereology, Akademiska University Hospital, Uppsala, Sweden, 16Danish Anaesthesia Allergy Centre, Allergy Clinic, Copenhagen University Hospital, Gentofte, 17Department of Hematology, Copenhagen University Hospital, Copenhagen, Denmark, 18Department of Internal Medicine I, Division of Hematology & Hemostaseology, Medical University of Vienna, Vienna, Austria, and 21Department of Medical Sciences, Hematology, Uppsala University, Uppsala, Sweden
Mastocytosis is a heterogeneous group of diseases defined by an increased number and accumulation of mast cells, and often also by signs and symptoms of mast cell activation. Disease subtypes range from indolent to rare aggressive forms. Mastocytosis affects people of all ages and has been considered rare; however, it is probably underdiagnosed with potential severe implications. Diagnosis can be challenging and symptoms may be complex and involve multiple organ-systems. In general it is advised that patients should be referred to centres with experience in the disease offering an individualized, multidisciplinary approach. We present here consensus recommendations from a Nordic expert group for the diagnosis and general management of patients with mastocytosis. Key words: mastocytosis; systemic mastocytosis; urticaria pigmentosa; classification; diagnostic criteria; mast cell; multidisciplinary management.
Accepted Dec 16, 2015; Epub ahead of print Dec 22, 2015
Acta Derm Venereol 2016; 96: XX–XX.
Sigurd Broesby-Olsen, Department of Dermatology and Allergy Centre, Odense University Hospital, Sdr. Boulevard 29, DE-5000 Odense C, Denmark. E-mail: sigurd.broesby-olsen@rsyd.dk
Mastocytosis is a heterogeneous group of diseases characterized by expansion and accumulation of tissue mast cells (MCs) in various organ systems (1, 2). Clinical manifestations are caused by MC infiltration and effects of mediators released from activated MCs. Mastocytosis has been considered rare, but is probably underdiagnosed and may affect children as well as adults.
This document has been prepared in a collaborative effort between existing mastocytosis centres in Denmark, Sweden, Norway and Finland and has developed through discussions at meetings in Stockholm, Vienna, Odense and London between 2010 and 2015. Furthermore, to adjust content and recommendations to internationally accepted guidelines the document has been critically revised by a major external discussant, the founder and coordinator of the European Competence Network on Mastocytosis (ECNM) (PV). Our aim is to provide general recommendations for healthcare professionals involved in diagnosing and treating patients with mastocytosis. Given the relative rarity and heterogeneity of the disease, which may have severe implications, it is generally advised that patients with mastocytosis are referred to centres with experience in the management and treatment of the disease, i.e. a centre of excellence of the ECNM (www.ecnm.net). Typically, these centres offer a multidisciplinary, coordinated and tailored approach to the individual patient.
WHO CLASSIFICATION AND DISEASE SUBTYPES
The WHO classifies mastocytosis into 7 subcategories, including cutaneous mastocytosis (CM), where major MC accumulations are limited to the skin, and systemic mastocytosis (SM), where internal organs are involved (2–4) (Table I). In general, most adults (> 95%) with mastocytosis are diagnosed with SM when fully investigated, with potential multiple clinical manifestations ranging from mild to very severe or even life-threatening (Fig.1).
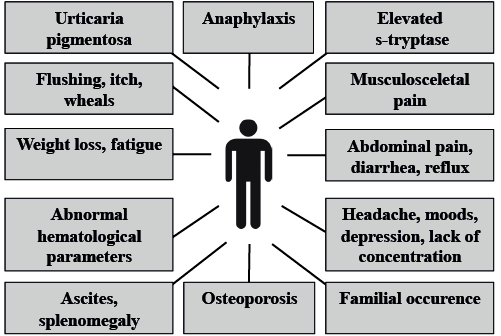
Fig. 1. Various clinical presentations of mastocytosis.
Table I. WHO classification of mastocytosis
|
Cutaneous mastocytosis (CM) |
|
Indolent systemic mastocytosis (ISM) |
|
Systemic mastocytosis with associated haematological non-MC-lineage disease (SM-AHNMD) |
|
Aggressive systemic mastocytosis (ASM) |
|
Mast cell leukaemia (MCL) |
|
Mast cell sarcoma (MCS) |
|
Extracutaneous mastocytoma |
From Horny HP, Metcalfe DD, Bennett JM, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008: 54–63.
In affected children, on the other hand, the disease manifestations are almost exclusively limited to the skin and symptoms are often mild. Whereas adult-onset mastocytosis is a chronic disease, paediatric mastocytosis has a tendency to spontaneous remission in approximately 70–80% of children in the teenage years (5, 6).
SM is divided into several subtypes, of which indolent SM (ISM) by far is the most prevalent (Table I). ISM is characterized by a stable or only very slowly progressing, benign course and a life-expectancy not differing from the (healthy) background population (7, 8). The main challenge in ISM is to control MC mediator-related symptoms. Subtypes of ISM include bone marrow mastocytosis (BMM), in which skin lesions are absent; and smouldering SM (SSM) where the burden of neoplastic MCs is high and clonal multi-lineage involvement is present. SSM is defined by the presence of at least 2 “B-findings” (Table II). Although organomegaly often occurs, organ damage is not present (2). In most patients with SSM the course remains indolent; however, it is advised to monitor patients regularly due to an increased risk for progression to advanced SM. Given its distinct pathogenesis and prognosis SSM can be regarded as a separate category of SM (2, 9).
Table II. WHO “B- and C-findings” in systemic mastocytosis
|
B-findings |
|
Bone marrow biopsy showing >30% infiltration by mast cells (focal, dense aggregates) and serum total tryptase level >200 ng/ml Signs of dysplasia or myeloproliferation in non-mast cell lineage, but insufficient criteria for definitive diagnosis of a haematopoietic neoplasm (associated non-MC-lineage clonal haematological disease; AHNMD), with normal or only slightly abnormal blood counts Hepatomegaly without impairment of liver function, and/or palpable splenomegaly without hypersplenism, and/or lymphadenopathy on palpation or imaging |
|
C-findings |
|
Bone marrow dysfunction manifested by one or more cytopaenia (absolute neutrophil count < 1.0 × 109/l, Hb < 10 g/dl, or platelets < 100 × 109/l), but no frank non-mast cell haematopoietic malignancy Palpable hepatomegaly with impairment of liver function, ascites and/or portal hypertension Skeletal involvement with large osteolytic lesions and/or pathological fractures* Palpable splenomegaly with hypersplenism Malabsorption with weight loss due to gastrointestinal mast cell infiltrates |
Smouldering systemic mastocytosis (SSM) requires that 2 or more B-findings and no C-findings are present. Aggressive systemic mastocytosis (ASM) requires presence of one C-finding and documentation of local mast cell (MC) infiltration. *Should not be confused with osteoporosis and resulting vertebral compression fractures, commonly seen in indolent systemic mastocytosis.
From Horny HP, Metcalfe DD, Bennett JM, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, et al, editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008: 54–63.
Mastocytosis in the skin (MIS) denominates patients with typical skin lesions in whom a full work-up for SM including a bone marrow (BM) investigation has not (yet) been performed (2). Most adult patients with MIS are considered to have ISM if investigated (10). However, there are adult patients with true CM, in whom the prognosis and organ involvement are different from those with SM. Therefore, a BM examination is recommended for all adult patients with MIS. In children, on the other hand, MIS in general does not lead to examination for SM; and in this situation CM is accepted as the final diagnosis (2).
The advanced forms of SM are much rarer than indolent forms and they almost exclusively occur in adulthood. Aggressive SM (ASM) is characterized by massive MC infiltration in organs such as bone marrow, liver and bone, resulting in organ damage (referred to as a “C-finding” (Table II). The prognosis in ASM is poor, but survival times vary depending on several different prognostic variables. One of the most important indicators of advanced disease is the percentage of MCs in BM smears. In ASM patients in whom the percentage of MCs in the bone marrow smears exceeds 5% the prognosis is grave, as many of these cases transform to mast cell leukaemia (MCL) within a short time. Therefore, these patients (5–19%) have been referred to as ASM in transformation (ASM-t) (11). As soon as the percentage of MCs in the BM smear increases to 20% the diagnosis changes to MCL.
SM-AHNMD is characterized by SM and the presence of an associated (concomitant) non-MC-lineage clonal haematological disease. The SM component is typically ISM or ASM, whereas the AHNMD component of the disease most often is a clonal myeloid neoplasm; a myelodysplastic syndrome (MDS), chronic myelomonocytic leukaemia (CMML), myeloproliferative disorder or acute myeloid leukaemia (AML); whereas lymphoproliferative disorders are far less common. The prognosis in SM-AHNMD depends both on the type of AHNMD and the type of SM and, in general, the treatment strategy is to treat the AHNMD- and the SM-part independently according to its own treatment guidelines (2, 4).
MCL is the rarest form of ASM, accounting for less than 1% of SM cases. The diagnosis is based on the presence of ≥ 20% of pathological MCs in the BM smear. In the classical variant, ≥ 10% of all peripheral leukocytes are MCs, whereas in the more frequent aleukaemic variant, MCs in the peripheral blood comprise less than 10% of all leukocytes. In general, MCL has a poor prognosis with a median survival time of less than one year. MCL can, however, be either acute or chronic. The acute form of MCL is characterized by the presence of C-findings (organ damage) and a rapid, malignant course, whereas the chronic form of MCL is defined by the absence of C-findings; and these patients may remain stable for longer, even though most patients with chronic MCL eventually transform to acute MCL and succumb to the disease (12).
The WHO classification further includes MC sarcoma and extracutaneous mastocytoma. These disease forms are exceptional rarities and will not be discussed further in the present article.
EPIDEMIOLOGY
Reports on epidemiological aspects of mastocytosis are scarce. In a recent nationwide study in Denmark based on Danish health registries an estimated prevalence of mastocytosis of all subtypes in adults was found to be 10 in 100,000 with a slight female predominance (8). In this study, indolent disease forms (ISM and CM) comprised 92%, SM-AHNMD 5%, ASM 2% and MCL less than 1% of cases, respectively. A similar prevalence of ISM was found in a single-centre study from Holland (13).
In general, the disease is highly likely to be underdiagnosed due to a lack of awareness among medical care providers and due to the very heterogeneous clinical presentation. With increasing awareness and new diagnostic options the prevalence is anticipated to increase.
The prevalence of paediatric mastocytosis is not known, but it is estimated to be higher than that of SM.
PATHOGENESIS
In most patients with SM a somatic gain-of-function point mutation in the KIT gene leads to a stem cell factor (SCF)-independent constitutive activation and autophosphorylation of KIT with subsequent expansion of clonal MCs. The cause of MC accumulation in mastocytosis apparently involves an upregulated recruitment of MCs from their progenitor cells and defective apoptosis in mature MCs rather than uncontrolled MC proliferation, since increased mitotic activity in neoplastic MCs is rarely encountered histologically, and furthermore, in the vast majority of patients, the disease follows a benign course.
By far the most frequent KIT mutation in SM involves codon 816, resulting in the replacement of an aspartic acid by a valine residue (D816V) that is critically involved in the kinase activity of KIT. Using highly sensitive methods this KIT D816V mutation can be detected in more than 90% of adults with mastocytosis, not only in lesional mast cells from the skin and bone marrow, but often also in circulating leukocytes in the peripheral blood (14–17).
The pathogenesis of paediatric mastocytosis is less well understood. As opposed to adult patients the KIT D816V mutation can be detected in only approximately 25–35% of affected children (6). KIT mutations in codons other than 816 are relatively more prevalent in the paediatric population compared with SM (6, 18); however, the mechanisms involved in the apparent spontaneous resolution observed in approximately 70–80% of children has not been elucidated. Other pathogenic factors appear to be responsible for the final disease phenotype in adult as well as paediatric mastocytosis because the burden of clonal MCs and presence of KIT mutations alone does not explain the remarkable heterogeneity of the clinical presentation and prognosis of the disease.
The complex physiology of the MC and its mediators is probably contributing to the varying clinical presentations and heterogeneous challenges in mastocytosis. The classic allergic immunoglobulin E (IgE)-mediated MC degranulation is one of the best described cellular reactions; however, MC activation is often non-IgE-mediated and can be induced by several different IgE-independent triggers. Furthermore, MCs may release numerous preformed as well as de novo synthesized mediators, of which histamine is the best known. Another well-known mediator is tryptase. Elevated baseline serum tryptase is characteristically found in SM, although a normal serum tryptase level does not rule out the presence of mastocytosis (2, 16). In the follow-up and monitoring of patients with mastocytosis s-tryptase is a widely used marker of MC burden (19). Furthermore, a rapid event-related rise and subsequent decrease in s-tryptase is of value when diagnosing MC degranulation in anaphylaxis (20, 21). Another group of MC mediators contributing to complex symptomatology are prostaglandins, of which prostaglandin D2 is best described in mastocytosis, and may be targeted by the use of low-dose aspirin, even though aspirin/non-steroidal anti-inflammatory drugs (NSAIDs) may also cause exacerbations in intolerant patients (22–24).
DIAGNOSTIC CRITERIA AND WORK-UP
Mastocytosis may come to clinical attention in several ways and be suspected as differential diagnosis to many conditions (Fig.1). Common clinical presentations include a spectrum ranging from gradually appearing typical skin lesions, allergic symptoms including anaphylaxis (e.g. after bee or wasp stings), unexplained gastrointestinal (GI) symptoms, headache, hypotension, neuropsychiatric symptoms, rheumatic symptoms/bone pain, osteoporotic fracture, to fatigue, cytopaenia, ascites and weight loss in advanced disease subtypes. Recognizing mastocytosis may be a challenge and diagnosis is often delayed due to lack of awareness among healthcare professionals.
In all adult patients and all childhood patients, a precise and complete physical examination is standard.
In paediatric mastocytosis the, by far, most common presenting symptom is the appearance of multiple skin lesions or a mastocytoma (25). In children the diagnosis of CM is largely clinical and a BM examination is not warranted or recommended except for the very rare situation where the child does not thrive and, in addition, shows persistently high and rising s-tryptase levels, abnormal blood count, or hepato-/splenomegaly.
Given the many clinical features and symptoms in adult mastocytosis that have to be recognized and handled by different specialists, a step-wise approach for initial work-up in adults with suspected mastocytosis is recommended (Fig. 2). The first step is a thorough skin examination for typical skin lesions, which may be inconspicuous and preferably should be confirmed by skin biopsy, especially in atypical cases. It is recommended that histological examination of skin biopsies should include staining for tryptase in addition to routine Giemsa and H&E stains, whereas CD25 and CD2 often is negative (26). If skin lesions are present the provisional diagnosis of MIS is established with recognition (and patient information) that this condition is a very strong indicator of the presence of SM in adults. In this situation, it is recommended to refer the patient for work-up in centres with experience in mastocytosis. In general, these centres recommend and perform a thorough BM examination in order to exclude or to establish the diagnosis of SM, to assess the BM MC burden, and to rule out or demonstrate the presence of another (associated) haematological disease.
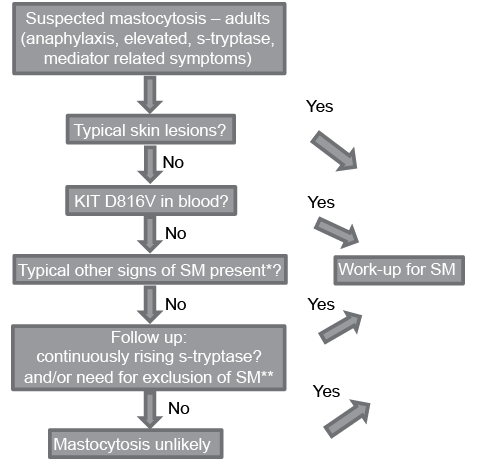
Fig. 2. Diagnostic algorithm presently used at Mastocytosis Centre Odense University Hospital and Oslo University Hospital. *Recurring spontaneous anaphylaxis, early osteoporosis, mast cell mediator related symptoms, GI symptoms, flushing **Haematological abnormalities, differential diagnostic purposes.
If skin lesions are not present, but s-tryptase levels are elevated and/or other signs and symptoms would argue for the presence of SM, it is recommended to perform a sensitive KIT D816V mutation analysis in the PB. If the KIT D816V mutation is detected a referral to a mastocytosis centre is recommended for work-up for SM regardless of s-tryptase level. If the mutation analysis is negative a further work-up for mastocytosis is warranted only if patient history and symptoms are highly indicative of mastocytosis and/or baseline s-tryptase is persistently above 20–30 ng/ml or continuously rising. If this is not the case, mastocytosis is unlikely and may, in practice, be ruled out. Of note, s-tryptase is not disease-specific for mastocytosis and may be elevated in healthy individuals, and in other conditions, such as chronic urticaria, kidney failure, chronic helminth infections, or other myeloid haematological diseases. A similar, step-wise approach, including KIT D816V testing in the algorithm is recommended by the ECNM (27).
The prerequisite for using this approach is the availability of a highly sensitive KIT D816V mutation analysis, since KIT D816V allele burden may be very low in ISM, often less than 0.01%, and hence there is a great risk of false-negative results if a less sensitive method is applied.
On the other hand, use of such a sensitive method may identify patients with SM among, for example, anaphylaxis patients not otherwise diagnosed due to absent skin lesions, lack of other MC mediator symptoms and normal baseline s-tryptase (28).
Diagnostic criteria for SM according to the WHO are listed in Table III. The diagnosis of SM requires investigation of an extracutaneous organ (tissue of choice: BM) and can be made when the major and at least one minor criterion or 3 minor criteria are found (3, 4).
Table III. WHO diagnostic criteria for systemic mastocytosis
|
Major criterion |
|
Multifocal, dense infiltrates of mast cells (15 or more mast cells in aggregates) detected in sections of bone marrow and/or other extracutaneous organ(s), and confirmed by tryptase immunohistochemistry or other special stains |
|
Minor criteria |
|
a. In biopsy sections of bone marrow or other extracutaneous organs, more than 25% of the mast cells in the infiltrate are spindle-shaped or have atypical morphology or, of all mast cells in bone marrow aspirate smears, more than 25% are immature or atypical mast cells b. Detection of KIT point mutation at codon 816 in bone marrow, blood or other extracutaneous organ(s) c. Mast cells in bone marrow, blood or other extracutaneous organs that co-express CD117 with CD2 and/or CD25 d. Serum total tryptase persistently >20 ng/ml* |
A diagnosis of systemic mastocytosis can be made if the major and one minor criterion or minimum 3 minor criteria are present. *Criterion not valid if there is an associated clonal myeloid disorder. From Horny HP, Metcalfe DD, Bennett JM, et al. Mastocytosis. In: Swerdlow SH, Campo E, Harris NL, et al. editors. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: IARC Press; 2008: 54–63.
The BM examination (with complete pathological work-up) in mastocytosis is challenging for the non-experienced pathologist. Therefore, it is recommended that the BM is examined by a pathologist with experience in diagnosing SM. Histopathological findings in BM sections in SM may be subtle and easily overlooked. Staining should include tryptase, CD117 and CD25, as well as CD30 if available, in addition to routine Giemsa and HE stains. Aberrant expression of CD2, CD25, and CD30 in neoplastic MCs can also be detected by flow cytometry, but this requires expertise. As part of the pathological evaluation the BM has to be evaluated thoroughly for the presence of an AHNMD, fibrosis, signs of ASM and signs of MCL. In a first step, a good-quality BM smear (or a touch preparation) needs to be investigated using Wright-Giemsa or May-Grünwald-Giemsa stain. In this investigation the most critical parameters are the percentage of MCs, the morphological grading of MCs, and the investigation of all other myeloid lineages, especially with regard to dysplasia, blast cell counts, and other signs of an AHNMD. In addition, chromosome analysis is standard; and in case of a suspected AHNMD, additional fluorescence in situ hybridization studies should be performed.
In adults diagnosed with SM further clinical work-up needs to be performed. In general, it is recommended that all adults are examined with a dual-energy X-ray absorptiometry (DXA) scan (see below) to exclude or confirm osteopaenia or osteoporosis. Furthermore, it is standard to perform a routine chest X-ray and ultrasound of the abdomen in order to define the spleen size and to exclude or demonstrate the presence of lymphadenopathy. A thorough allergological work-up in case of anaphylaxis is important. Depending on symptoms and findings in the individual patient, further work-up may include GI endoscopy and imaging, radiological bone examinations, neuropsychological evaluation and other investigations relevant for ruling out differential diagnoses.
CLINICAL MANIFESTATIONS AND TREATMENT OPTIONS
General considerations
In general, patients with mastocytosis are best handled by centres with experience in managing the disease, using a coordinated, multidisciplinary approach and it is advised that patients with mastocytosis are referred to such a centre for evaluation, if possible.
Treatment and follow-up should be focused on the individual patient, and tailored to patient’s symptoms, disease manifestations, and the clinical course. Treatment options can be divided into anti-mediator therapy, aimed at reducing release or effects of MC mediators, and cytoreductive therapies aimed at reducing MC burden. Due to potential side-effects of currently available drugs, and given the normal life expectancy in ISM and CM, the latter should be reserved for the few patients with advanced SM or ISM with high MC burden and severe treatment-resistant symptoms. A thorough provision of patient information and counselling is standard. Most patients seek information through the Internet, but this may vary in quality and relevance and thus may lead to misinformation and unnecessary concerns. The ECNM homepage provides updated information to patients and doctors, as well as list of centres of excellence and of reference centres in most European countries.
Identifying and avoiding relevant triggers of MC degranulation is of importance in all patients. Although some triggers are commonly reported (e.g. heat, insect stings, alcohol, friction, spicy foods), general advice to avoid a long list of theoretical triggers is not helpful, but must be individualized and related to the patient’s experience. Patients should, however, be informed about potential risk situations (e.g. wasp sting, contrast media, general anaesthesia), and it is advisable to provide written information to the patient and involved healthcare personnel.
Most centres advise that all adults with mastocytosis and children with severe disease are equipped with an adrenaline auto-injector for use in case of anaphylaxis (see below), although firm evidence supporting this approach in clinical trials is lacking.
Skin manifestations
The most common presenting symptom in mastocytosis, especially in children, is the gradual onset of pigmented, reddish-brown, maculopapular well-demarcated skin lesions traditionally named urticaria pigmentosa (UP) (Fig. 3). However, although the term UP is still widely used, the more correct term for skin lesions in adult mastocytosis is MIS, as explained above. In children and in adult cases where SM has been ruled out, skin lesions are termed maculopapular CM (MPCM) (3). The number of skin lesions may vary from very few (<5), easily missed in routine clinical examination, to widespread skin involvement covering most of the skin. In adults skin lesions are typically monomorphic and small-sized (up to 5 mm in diameter), whereas lesions are often larger and more polymorphic in paediatric mastocytosis (26). Skin lesions typically show a wheal-and-flare reaction upon rubbing or stroking; so-called Darier’s sign (Fig. 4). In daily clinical practice a useful approach to elicit this sign is applying 5 strokes with a tongue spatula with a moderate pressure over the skin lesions, and waiting 3–5 min (up to 15 min) to evaluate response. False-negative results are sometimes encountered due to intake of antihistamines and individual thresholds. Dermographism (stripes of whealing in the normal skin upon scratching) may be seen in mastocytosis, but is also a common finding in some types of urticaria, and should not be confused with Darier’s sign.
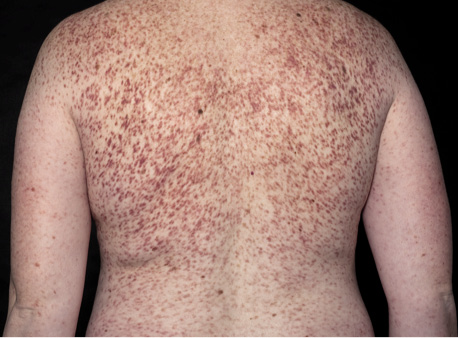
Fig. 3. Typical mastocytosis skin findings in an adult patient with symmetrically distributed, small, partly confluent, macular, brown, monomorphic lesions, traditionally termed urticaria pigmentosa, on the trunk and arms.
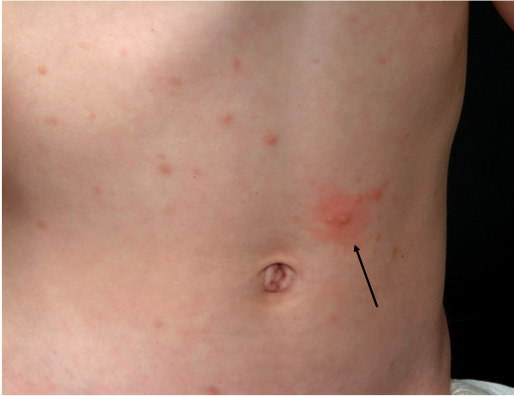
Fig. 4. Darier’s sign is an important dermatological finding and may be elicited by stroking a mastocytosis skin lesion approximately 5 times with moderate pressure with a tongue spatula. Within a few minutes, a wheal and flare reaction of the lesion develops (arrow).
The presence of telangiectasia, along with MPCM-like skin lesions, has traditionally been termed telangiectasia macularis eruptiva perstans (TMEP). There is, however, no clear evidence that this is a separate subtype of CM, or whether it merely represents a well-vascularized MPCM with dilated vessels in the skin. Therefore, even though telangiectasia may sometimes be a predominant clinical feature, the term TMEP should no longer be used (26).
In children the skin lesions are often clinically larger (Fig. 5) and polymorphic. Other subtypes include mastocytoma of the skin and diffuse cutaneous mastocytosis (DCM) (see below).
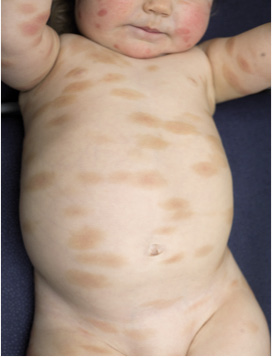
Fig. 5. Maculopapular cutaneous mastocytosis (MPCM) in a child with larger, reddish-brown lesions of different sizes over the trunk.
Skin symptoms include pruritus, flushing and whealing, often triggered by physical stimuli such as heat, cold, sunlight, and friction. These MC mediator-related skin symptoms can often be controlled by the use of H1-anthistamines that may be administered in increasing doses and up to 4 times daily in line with treatment principles in chronic urticaria.
Cosmetic complaints are a major issue in many patients, especially in younger adults, and should not be underestimated. The challenge of treating the skin lesions is, however, that they are caused by MC infiltration in the skin, rather than MC mediators, and hence require reduction in this infiltration. Indeed, the disappearance of skin lesions has been reported after the use of cytoreductive therapies, such as cladribine and tyrosine kinase inhibitors, in patients with ASM. However, use of potentially toxic drugs in ISM or CM is not warranted for cosmetic indications. Therefore, current studies aim at identifying KIT-targeting tyrosine kinase inhibitors that are less toxic and can be applied safely in these patients. At present, however, treatment targeting the cosmetic burden of skin lesions is limited to ultraviolet (UV) therapy and short-term high-potency topical steroids under plastic occlusion, alone or in combination. These treatments should be used with caution due to potential cutaneous side-effects, but may, in addition to reducing MC mediator skin symptoms, also make skin lesions somewhat less visible, although the effect is temporary. A few reports exist on the effects of laser treatment for skin lesions, e.g. on the face; however, experience is limited.
Anaphylaxis
The prevalence of anaphylaxis in the general population has been estimated to be 0.05–2%, whereas reported prevalence in adults with mastocytosis is 35–50% and 5–10% in children with this disease. Common elicitors include Hymenoptera sting (approximately 50%), drugs including opioids, contrast media, NSAIDs and muscle relaxants, whereas more infrequent triggers are IgE-mediated allergies to foods, antibiotics and latex (29). More general triggers, such as physical stress, exercise, fever, alcohol, cold/heat and spicy foods, are not common causes of severe anaphylaxis by isolated exposure, but may act in combination. Other triggers may be relevant in the individual patient, and each patient with mastocytosis should be advised to record all events and to learn (as far as possible) what triggers are harmful, in order to avoid these. In approximately one-third of anaphylaxis patients with mastocytosis no obvious cause can be identified even after thorough evaluation, so-called idiopathic anaphylaxis, probably reflecting a spontaneous MC activation (29, 30).
In general, the incidence of IgE-mediated allergic rhinitis, asthma and food allergies is not more frequent in mastocytosis, but symptoms may be more severe and difficult to control when the conditions co-exist.
No firm predictors or surrogate markers have been identified to allow an individual risk assessment for anaphylaxis in mastocytosis and further studies are needed. In paediatric mastocytosis the risk may be correlated with the severity and character of skin lesions as well as the level of s-tryptase. In contrast, risk of anaphylaxis in adults does not correlate with MC burden or may even correlate inversely, and adult patients without skin lesions (BMM) may even be more prone to anaphylaxis than patients with typical ISM. In addition, patients with ASM and MCL may paradoxically lack MC mediator-related symptoms.
Presently, even though firm evidence is lacking, most centres recommend that all adults with mastocytosis are equipped with an adrenaline auto-injector for use in case of anaphylaxis, whereas in paediatric mastocytosis only children with very extensive skin involvement, bullous skin reactions, persistently high tryptase levels (> 100), or previous anaphylaxis need to be prescribed an adrenaline pen. In addition, patients should be informed about potential MC triggers and risk situations, as described above, and a thorough evaluation and allergological work-up is recommended in case of anaphylaxis. Patients with bee/wasp (Hymenoptera) allergy may be offered venom immunotherapy (VIT) that must be continued life-long. Due to risk of severe side-effects of venom injections it is advised that VIT in mastocytosis is performed by specialized allergy centres with experience in mastocytosis.
Gastrointestinal symptoms
GI symptoms, including abdominal pain, cramping, diarrhoea and peptic symptoms are frequently encountered in SM (29, 31). Symptoms may be severe and/or have overlap with symptoms of other GI diseases (gastroesophageal reflux, Helibacter pylori associated GI ulcers, irritable bowel syndrome, inflammatory bowel diseases, coeliac disease, malignant disease). Therefore, these patients must be evaluated to rule out these differential diagnoses. The pathogenesis of GI symptoms in mastocytosis involves the accumulation of clonal MCs in the GI tract, as well as MC mediator release, which may be triggered by various stimuli, e.g. spicy foods, alcohol, stress, or even occur spontaneously. Histology of GI biopsies may exhibit infiltrates of neoplastic MCs; however, changes can be focal and subtle, making recognition challenging, especially compared with normal GI mucosa (32). Reported, characteristic endoscopic findings in GI-mastocytosis include mucosal erythema, granularity or nodularity; however, approximately half of affected patients do not show endoscopic abnormalities in spite of histological changes and it is advisable to take multiple random biopsies at different sites in addition to visibly abnormal areas (31–35). Treatment options include H2 blockers, proton-pump inhibitors and oral sodium cromolyn that may be combined. Individual triggers for symptoms should be identified by case history and avoided if possible. In a minor subset of patients a true IgE-mediated food allergy may be suspected needing specialized allergological work-up. In the rare aggressive subtypes of SM, malabsorption and weight loss may occur due to severe MCs infiltration in the GI tract. In this situation cytoreductive therapy may be warranted (36, 37).
Bone/osteoporosis
Within all subcategories of SM, more than half of patients show skeletal involvement, including not only osteoporosis and osteolytic lesions, but also osteosclerosis (38, 39). Osteopaenia and osteoporosis are the most frequent and important bone complications of SM. Approximately 30% of adult patients with SM have osteoporosis, which may go undetected due to lack of symptoms (40). The pathogenesis involved has not been fully elucidated but may involve MC-derived cytokines, receptor activator of nuclear factor kappa-B ligand (RANKL), as well as effects of MC mediators (proteolytic enzymes and heparin) on bone metabolism and remodelling (41, 42). Onset may be early in life and no clear predictors have been identified, thus it is advised to screen all adult patients with SM by DXA. Further work-up and radiological examinations, including total spine radiography, depend on individual clinical presentation, including the presence of focal skeletal symptoms. Patients with severe, atypical or progressive osteoporosis and young individuals should be referred to a specialist in osteoporosis for evaluation. Clinical trials on specific treatments in SM-associated osteoporosis are lacking. Therefore, at present it is advised to follow general guidelines for osteopaenia and osteoporosis concerning treatment and follow-up, including vitamin D and calcium supplements and bisphosphonates (39). Early institution of bisphosphonates, as soon as the T-score on DXA is below –2, has been suggested in a previous consensus document (2). The effectiveness of new drugs, such as denosumab, remains to be determined. Based on our limited understanding of the pathogenic mechanisms underlying the development and evolution of osteoporosis in SM, no more specific treatment or prophylaxis can be recommended presently. It is probable that MC mediators are involved and therefore it may be hypothesized that an effective anti-mediator treatment may prevent development of osteoporosis; however, evidence supporting this approach is lacking. On the other hand, it appears that MC burden does not correlate with increased frequency of osteoporosis and cytoreductive treatments are generally not warranted for this indication (43, 44). It must be stressed that the presence of an osteoporotic fracture is not a sign of aggressive disease, i.e. a C-finding, as opposed to the rarely encountered large, osteolytic bone lesions that can sometimes be detected in patients with ASM or MCL (2).
Musculoskeletal involvement
Poorly localized pain from soft tissue and bone is a frequent complication of SM and probably related to effects of MC mediators including prostaglandins, but the pathogenic mechanism is not well understood (24, 44, 45). Laboratory tests, radiological imaging and referral to a rheumatologist may be relevant to rule out differential diagnoses. Therapeutic options include conventional analgesics, NSAIDs/aspirin if tolerated, physiotherapy and exercise. Furthermore, blocking MC mediators may aid in gaining control of symptoms (46, 47). Opioids should, in general, be avoided given the chronicity of SM, and due to side-effects including potential MC triggering.
Neuropsychiatric symptoms
Neuropsychiatric symptoms are frequently encountered in mastocytosis and include headache, depression, anxiety, and cognitive impairment and memory problems (48–52). It is estimated that these types of symptoms affect approximately one-third of patients with SM and may sometimes be severe (44). The mechanism is not clear, but may involve the effect of MC mediators in the central nervous system. Other factors, such as difficulty coping with a chronic disease such as mastocytosis, may also play a role. These symptoms may have a severe impact on daily life in many SM patients and therefore deserve recognition. As is the case for many of the manifestations of mastocytosis a thorough search for differential diagnoses is important and includes a psychiatric and/or neurological work-up, depending on the type and severity of symptoms. Treatment options comprise blocking of MC mediators by H1-antihistamines, leukotriene antagonists, antidepressants and psychological support. Other potentially useful drugs could include sodium cromolyn and omalizumab (53–55); however, studies on this manifestation of SM and treatment options are required.
ADVANCED FORMS OF SYSTEMIC MASTOCYTOSIS
Aggressive systemic mastocytosis (ASM) is characterized by organ dysfunction due to MC infiltration. The diagnosis of ASM requires the presence of at least one C-finding (Table III) related to a documented local MC infiltration. Thus, biopsies from, for example, liver or spleen may be required to document the causal relationship with a malignant MC-infiltrate and thus the presence of a C-finding. Anaemia, thrombocytopenia, neutropaenia, ascites, hypoalbuminaemia, and considerable weight loss are frequently present in patients with ASM. Large osteolysis with and without pathological fractures may also be detected.
In ASM cytoreductive treatment is needed in order to reduce the MC burden and restore organ function, whereas MC mediator-related symptoms often play a minor role. Usually, the slowly progressing form of ASM can be kept under control with interferon-α or chlorodeoxyadenosine (cladribine) for months or even years (56). If these treatments fail, PKC412 (midostaurin), an oral multi-kinase inhibitor blocking wild-type as well as mutated KIT, should be considered, as it has been demonstrated that this drug can induce clinically meaningful responses in most patients with ASM. In addition, PKC412 has been described to have an effect on overall survival in patients with ASM and MCL (57). The tyrosine kinase inhibitors nilotinib and dasatinib, which are inhibitors of mutated KIT, have shown only minor clinical effects, whereas imatinib has no therapeutic role in the presence of the KIT D816V mutation, but only in patients without this mutation (9). In patients with rapidly progressing ASM, ASM in transformation to MCL (ASM-t), or overt MCL, more intensive therapy is usually required. However, MCL can be divided into acute and chronic forms. In acute MCL cladribine or PKC412 have only limited effect, whereas patients with the less aggressive chronic MCL have a longer survival when treated with cladribine or midostaurin (12). In younger patients with a suitable donor, allogeneic stem cell transplantation (SCT) is the only treatment with a chance of inducing stable remission in patients with rapidly progressing ASM or acute MCL (9, 58). In these patients, polychemotherapy (as otherwise given to patients with high-risk AML) is initially administered. One standard protocol is fludarabine, cytarabine and granulocyte colony-stimulating factor (FLAG). In those who show a good response, 2–3 cycles are given, and the patient is then prepared for SCT.
In SM-AHNMD treatment should be guided by both the ASM component and the non-MC neoplasm. In those with ASM-AHNMD, treatment often needs to be adjusted to the ASM-component of the disease. By contrast, in those with ISM-AHNMD, the cytoreductive therapy follows the guidelines for treating the AHNMD type (as if no SM had been diagnosed).
SPECIAL ASPECTS
Paediatric mastocytosis
The most common clinical scenario leading to diagnosis of mastocytosis in the paediatric population is a child presenting with skin lesions within the first 2 years of life. Symptoms in paediatric mastocytosis are usually mild to moderate and are often limited to the skin (25, 59, 60). It is, however, recommended that children with mastocytosis are offered an evaluation by experienced mastocytosis centres with established collaboration with paediatricians.
An as-yet unsolved enigma is the spontaneous resolution observed in the majority of children as described above (5). One possible explanation is differences in mutational profiles compared with adults. In the paediatric population it is only possible to detect the KIT D816V mutation in approximately 25–35%, while 80–95% of adult patients with typical ISM carry this mutation (6). Apparently non-D816V KIT mutations are more frequent in children (18).
The clinical character of skin lesions in paediatric mastocytosis is more heterogeneous than in adults. Some children exhibit classic, small monomorphic UP-skin lesions like adults, whereas others have larger, polymorphic lesions (61) (Fig. 5). Recent data have suggested that children presenting with monomorphic small-sized lesions have a higher risk of having persistent mastocytosis, whereas in those with polymorphic larger lesions, the disease more often resolves before or at puberty (26, 62).
A rare paediatric subtype is diffuse cutaneous mastocytosis (DCM) in which the whole integument is infiltrated by MCs. Children with DCM do not present with individual lesions, but rather exhibit generalized erythema, usually with thickened, darker skin and a very pronounced dermographism with often bullous reactions. The clinical presentation of DCM may be dramatic, with extensive blistering and hypotensive episodes; however, improvement is usually seen within the first 3–4 years. Although tryptase levels are often increased at presentation, evidence of systemic involvement is usually not found and spontaneous regression over time is the rule (26, 63).
Another type of CM almost exclusively seen in the paediatric population is mastocytoma of the skin, often presented in clinic by worried parents as an itchy and swollen naevus. Even though the vast majority of patients have very limited symptoms that do not require treatment, occasionally these may be accompanied by generalized flushing and hypotensive episodes, i.e. anaphylaxis, for which excision may be considered (25). Symptomatic treatment options include H1-anthistamines, topical or intralesional steroids.
A diagnosis of CM and not SM is presumed in all children with typical MPCM-skin lesions and a BM examination is not recommended except for the very rare situation where the child does not thrive, and in addition shows persistently high and rising s-tryptase, abnormal blood counts, or hepato/splenomegaly. A follow-up is recommended, and if the disease persists into adulthood a work-up as described for adults, including a BM examination, should be performed (2). Aggressive subtypes of mastocytosis are exceedingly rare in the paediatric population.
At present no firm predictors for persistent disease exist. From a biological angle it is tempting to assume that children carrying the KIT D816V mutation will persist into adult mastocytosis. However, long-term prospective studies have not been performed, and there are children with KIT D816V-positive skin lesions in whom these lesions disappear during puberty. Similarly, disease onset after 3 years of age may be predictive of persisting disease. Finally, the clinical appearance of small monomorphic UP-skin lesions, i.e. skin lesions like those found in adults, are an assumed clinical predictor for persistence in patients with childhood mastocytosis (62).
Treatment should be aimed at symptoms and is not always needed. Information about common triggers for MC activation, such as friction, heat and cold, should be given, but should be individualized and, apart from physical stimuli to the skin, most other theoretical triggers do not have clinical relevance in childhood mastocytosis and it must be stressed that it is not helpful to provide a long list of potential MC triggers to avoid. To that end, counselling and thorough information for often worried parents is of great help, bearing in mind that information found on the Internet on this very heterogeneous disease may vary greatly in relevance and quality and can thereby lead to misinformation and unnecessary concerns.
Antihistamines in general suffice to control skin symptoms. Non-sedating H1-antihistamines should be prescribed. Short periods of sedating antihistamines may be warranted for sleep-disturbing symptoms, but should not be prescribed for long-term use. Topical steroids may dampen skin symptoms and increase the threshold for MC activation, but should be prescribed only for short-term use due to potential side-effects in the skin (25).
Risk of anaphylaxis in paediatric mastocytosis is much lower than in adult mastocytosis, and presently it is only warranted to prescribe an adrenaline pen as emergency medication for children with very extensive skin involvement, bullous skin reactions, persistently high tryptase levels (>100) or previous anaphylaxis (64–66).
ANAESTHESIA
The incidence of perioperative anaphylaxis in patients with mastocytosis is not known. However, it is generally assumed that the perioperative setting is a high-risk environment for mastocytosis patients due to the combination of multiple drug administrations and exposure to potential triggers of mast degranulation, such as emotional stress, cooling/heating and pressure from tourniquets. Such assumptions have led to generalizations regarding drug groups to avoid when performing general anaesthesia in mastocytosis patients, which in some cases have led to suboptimal care, i.e. with regard to management of postoperative pain. There is very limited literature on the subject and most recommendations are therefore based on the lowest level of evidence, such as information from case reports combined with expert opinion. Individual reports of reactions in patients with mastocytosis largely implicate drugs such as opioids, NSAIDs, antibiotics, radio-contrast media, local anaesthetics and muscle relaxants reflecting drugs commonly used and also implicated in perioperative anaphylaxis in non-mastocytosis patients. Reports are presenting conflicting information, and affirmative evidence for avoidance of specific drugs is therefore lacking.
There are no studies of adult patients and, to date, very few studies of the perioperative management of paediatric mastocytosis patients have been published. These point in the direction that, overall, anaesthesia is safe in the majority of paediatric mastocytosis patients without imposing generalized restrictions or precautions (67, 68).
Although the general risk of anaphylaxis in adult mastocytosis patients is greater than in children, it is still considered to be low in the majority of patients. Therefore, an individualized risk assessment should be carried out before surgery and anaesthesia in both adults and children with mastocytosis.
Patients without previous episodes of anaphylaxis, perioperatively or to drugs, require no special precautions other than continuing usual anti-mediator therapy.
Patients with previous episodes of drug allergy should avoid specific drugs suspected of eliciting reactions. If previous reactions occurred perioperatively specialized allergological investigations, taking into account all perioperative exposures including anaesthetic drugs, latex, disinfectants, dyes, and antibiotics, should be carried out to try to identify the eliciting drug.
Procedures performed in regional (epidural/spinal), topical or local anaesthesia are generally considered safe (69–71) and precautions are warranted only in patients with a prior reaction.
In general, it is advised that mastocytosis patients inform the anaesthesiologist and the surgeon in good time preoperatively, to allow information to be gathered from previous anaesthesia records, so that previously tolerated anaesthesia regimens can be re-used. Detailed planning of the anaesthetic and the management of postoperative pain may be useful to avoid confusion and discussions about which drugs may be used on the day of surgery.
Some centres advocate a baseline s-tryptase prior to surgery so it is ready for comparison if a perioperative reaction occurs and s-tryptase is sampled at the time of reaction (68).
The use of premedication is not evidence-based, and no clear consensus exists, but is widely practiced. The patient’s regular anti-mediator therapy should be continued on the day of surgery and may be supplemented with premedication with a combination of antihistamines (H1 and H2 antagonists) and glucocorticoids (72).
For all mastocytosis patients attention should be paid to reducing/avoiding triggers, such as emotional stress, excessive cooling/heating, pain, friction, prolonged pressure from tourniquets and other mechanical factors. Premedication with benzodiazepines is advocated in some centres to reduce anxiety and emotional stress.
When anaesthetizing a patient with mastocytosis the anaesthesiologist should be alert for early signs of anaphylaxis, such as flushing and urticaria (which may also be absent or not easily noticed), hypotension, tachycardia and bronchospasm and be prepared to treat life-threatening symptoms with adrenaline according to international guidelines.
VENOM IMMUNOTHERAPY
Patients with IgE-mediated anaphylaxis to Hymenoptera should be offered venom immunotherapy (VIT) to honeybee or wasp or both (73). This treatment reduces the risk of life-threatening anaphylactic episodes and should be performed life-long in patients with mastocytosis. VIT in mastocytosis patients should be performed by experienced allergy centres due to the risk of severe side-effects, including anaphylaxis, and the need for dosage adjustments and concomitant treatments.
PREGNANCY
In general, pregnancy in mastocytosis is uncomplicated as long as physicians are aware of the presence of the underlying disease. Approximately 25–33% of patients experience some worsening of their mediator-related symptoms during pregnancy, whereas 10–25% of patients experience some relief of their mediator-related symptoms (74). Labour and delivery is, in general, uncomplicated without maternal or foetal complications and, overall, there is no need for special precautions/observations (see anaesthesia above). Anti-mediator treatment of safe type, such as cetirizine or fexofenadine in normal dosages, should be continued during pregnancy and labour (75).
The authors declare no conflicts of interest.
Contact information for Nordic mastocytosis centres can be found at: www.mastocytose.dk.
REFERENCES

