


1Department of Dermatology, 2Division of Skin Surface Sensing, Department of Dermatology, Kyushu University, 3Research and Clinical Center for Yusho and Dioxin, Kyushu University Hospital, Kyushu University, Fukuoka, and 4Department of Dermatology, St Marianna University School of Medicine, Kanagawa, Japan
Plaque psoriasis and pustular psoriasis are overlapping, but distinct, disorders. The therapeutic response to biologics supports the pivotal role of the tumour necrosis alpha (TNF-α)/ interleukin (IL)-23/IL-17/IL-22 axis in the pathogenesis of these disorders. Recently, functional activation of the IL-36 receptor (IL-36R) was discovered to be another driving force in the pathogenesis of psoriasis. This was first highlighted by the discovery that a loss-of-function mutation of the IL-36R antagonist (IL-36Ra) causes pustular psoriasis. Although the TNF-α/IL-23/IL-17/IL-22 axis and the functional activation of IL-36R are fundamentally involved in plaque psoriasis and pustular psoriasis, respectively, the 2 pathways are closely related and mutually reinforced, resulting in full-blown clinical manifestations. This review summarizes current topics on how IL-36 agonists (IL-36α, IL-36β, IL-36γ) signal IL-36R, the pathological expression of IL-36 agonists and IL-36Ra in plaque and pustular psoriatic lesions, and the cross-talk between the TNF-α/IL-23/IL-17/IL-22 axis and the functional activation of IL-36R in the epidermal milieu.
Key words: psoriasis; pustular psoriasis; IL-36; IL-36 receptor; IL-36 receptor antagonist; IL-17; IL-22.
Accepted Oct 2, 2017; Epub ahead of print Oct 2, 2017
Acta Derm Venereol 2018; 98: XX–XX.
Corr: Masutaka Furue, Department of Dermatology, Kyushu University, Maidashi 3-1-1, Higashiku, Fukuoka, 812-8582, Japan. E-mail: furue@dermatol.med.kyushu-u.ac.jp
Psoriasis is a common, immune-mediated, chronic inflammatory, erythemato-desquamative skin disease that is characterized by the altered proliferation and differentiation of keratinocytes with infiltration of neutrophils, dendritic cells (DCs) and T cells (1, 2). Psoriasis has shown a diverse prevalence across populations worldwide: 2.5% in Europeans, 0.05–3% in Africans and 0.1–0.5% in Asians (1, 2). Psoriasis is divided into 2 major clinical variants, plaque psoriasis and generalized pustular psoriasis, with shared pathogenetic backgrounds (1–4). Pustule formation of plaque psoriasis is not uncommon in severe cases (1, 4). A recent study revealed that palmoplantar pustulosis, a localized variant of pustular dermatosis, shares a similar pathogenesis with the generalized form (5). The therapeutic guidelines include the use of topical steroids, topical vitamin D, systemic immunosuppressants and various biologics, such as anti-tumour necrosis factor (TNF) α, anti-interleukin (IL)-23 and anti-IL-17 antibodies (6–14). However, treatment adherence and the quality of life of the affected patients are generally very low (1, 15–20). Recent genome-wide association studies have identified many susceptibility loci for psoriasis and pustular psoriasis, including HLA-C*06:02, LCE3D, IL23R, CARD14 and IL36RN (21–24). These susceptibility genes are predominantly related to the innate and adaptive immune systems and to skin barrier functions (1, 21).
Psoriasis often coexists with other systemic diseases, such as arthritis, diabetes mellitus, arterial hypertension, obesity and cardiovascular diseases (the so-called “psoriatic march” or “inflammatory march”) (25–30). Psoriasis is also co-morbid with other autoimmune diseases, including Hashimoto’s thyroiditis and autoimmune bullous diseases (31–36). The recent therapeutic success of anti-TNF-α, anti-IL-23 and anti-IL-17 antibodies, as well as the diverse action of IL-22, has emphasized the pivotal role of the TNF-α/IL-23/IL-17/IL-22 pathway in the pathogenesis of plaque psoriasis and pustular psoriasis (2, 37, 38). In addition to the TNF-α/IL-23/IL-17/IL-22 pathway, IL-36 and IL-36 receptor antagonist (IL-36Ra/IL36RN) are currently the focus of much attention. The gene and immunohistological expression of IL-36 members is upregulated in plaque psoriasis and pustular psoriasis (39–41). Moreover, IL36RN deficiency has been associated with the development of generalized pustular psoriasis (23, 42). This review summarizes recent topics on IL-36 signalling with reference to plaque psoriasis and pustular psoriasis.
Epithelial cells, including keratinocytes, are a good source of the IL-1 family (IL-1F), which is composed of 11 members: IL-1α, IL-1β, IL-1 receptor antagonist (IL-1RN), IL-18, IL-33, IL-36α/IL-IF6, IL-36β/IL-1F8, IL-36γ/IL-1F9, IL-36Ra/IL-1F5, IL-37/IL-1F7 and IL-38/IL-1F10 (43–45). IL-36α, IL-36β, IL-36γ and IL-36Ra use the same receptor, IL-36R (previously called IL-1Rrp2 or IL-1RL2), coupled with the IL-1 receptor accessory protein (IL1RAP), which is a common subunit shared with IL-1R and IL-33R (40, 44). IL-36α, IL-36β and IL-36γ show similar levels of agonistic activity upon binding to IL-36R (44). However, ligation of IL-36R by IL-36Ra does not induce the recruitment of IL1RAP and therefore does not initiate a signalling response (44, 45).
Like other IL-1 family members, such as IL-1β and IL-18, proteolytic processing is required to unleash the proinflammatory activity of IL-36α, IL-36β and IL-36γ (4, 46). IL-36α, IL-36β and IL-36γ are processed and activated differentially by the neutrophil granule-derived proteases cathepsin G, elastase, and proteinase-3, increasing their biological activity by approximately 500-fold (46). For endogenous processing, caspase 1 is essentially involved in gene expression, and caspase 3 is involved in the release of IL-36γ (47). Other endogenous processing pathways of IL-36α, IL-36β and IL-36γ are not yet fully understood. However, a recent study by Ainscough et al. (48) demonstrated that keratinocyte- or fibroblast-derived cathepsin S is a key skin-resident enzyme that cleaves full-length IL-36γ into the active form IL-36γ-Ser18. It is feasible that IL-36γ-Ser18 per se induces hyperkeratosis and CXCL8 (IL-8) production in a skin-equivalent model (48). In addition to CXCL8 production, IL-36γ upregulates the production from human keratinocytes of CXCL1 (Gro1), CXCL10 (IP10) and CCL20 (MIP3A), which is differentially mediated by p38 MAPK, JNK MAPK and NF-κB, respectively (49).
Human keratinocytes apparently express IL-36α, IL-36β, IL-36γ, IL-36Ra and their receptor IL-36R/IL1RAP (4, 50). The treatment of human keratinocytes with IL-36α, IL-36β or IL-36γ upregulates the gene expression of various chemokines for macrophages (CCL3, CCL4, CCL5, CCL2, CCL17 and CCL22), T cells (CCL20, CCL5, CCL2, CCL17 and CCL22), and neutrophils (CXCL8, CCL20 and CXCL1). In addition to keratinocytes, human monocytes and myeloid DCs, but not CD4+ or CD8+ T cells, express IL-36R, while all subsets express IL1RAP. IL-36R was not detected on activated T cells stimulated with anti-CD3/CD28 antibodies. The circulating neutrophils also lacked IL-36R and did not respond to IL-36 treatment (50).
Myeloid DCs express approximately 10 times higher IL-36R levels than monocytes (50). Treatment with IL-36α, IL-36β or IL-36γ significantly enhances the surface expression of CD83, CD86 and HLA-DR, as well as the release of IL-1β and IL-6 in DCs. In addition, the surface expression of IL-36R is augmented during maturation of monocyte-derived DCs cultured with IL-4 and granulocyte-macrophage colony-stimulating factor. IL-36α, IL-36β or IL-36γ further accelerates the phenotypic and functional maturation of monocyte-derived DCs (50). Human M2 macrophages and Langerhans cells are also target cells of IL-36 signalling to produce IL-6 (51). These studies suggested that IL-36 members released from keratinocytes could feasibly induce a plethora of chemokines in keratinocytes in an autocrine fashion, recruiting circulating myeloid DCs, monocytes, T cells and neutrophils to the skin. IL-36 members also support the phenotypic and functional maturation of antigen-presenting cells (DCs and monocytes), which mediate local inflammation.
The pathogenetic importance of “functional activation of IL-36R” in psoriasis and pustular psoriasis was first proposed by findings that missense mutations in IL36RN were associated with pustular psoriasis (23, 52). Elevated levels of IL-36Ra are believed to counteract the proinflammatory reaction of IL-36 in plaque psoriasis and pustular psoriasis (4, 41). Therefore, loss of function mutation of IL36RN exaggerates IL-36 signalling (23, 52). Keratinocytes from patients with IL36RN mutation produced higher levels of CXCL8 than keratinocytes from controls in response to IL-36α, IL-36β and IL-36γ, as well as to IL-1β or polyinosinic-polycytidylic acid (poly [I:C]), which is a synthetic ligand specific for Toll-like receptor 3 (TLR3) (23). Mutations in IL36RN have been shown to account for 46–82% of cases of pustular psoriasis without associated plaque psoriasis (42, 53). A recent meta-analysis of 233 generalized pustular psoriasis cases revealed that carriage of 1 or 2 IL36RN mutant alleles conferred a more severe clinical phenotype with an earlier age of onset and an increased risk of systemic inflammation compared with non-carriage (54). A gene-dosage effect is also apparent because homozygous carriers have an earlier age of onset than heterozygotes (4, 54). These studies have emphasized the importance of gene mutations of IL36RN and subsequent “functional activation of IL-36R” in the pathogenesis of pustular psoriasis.
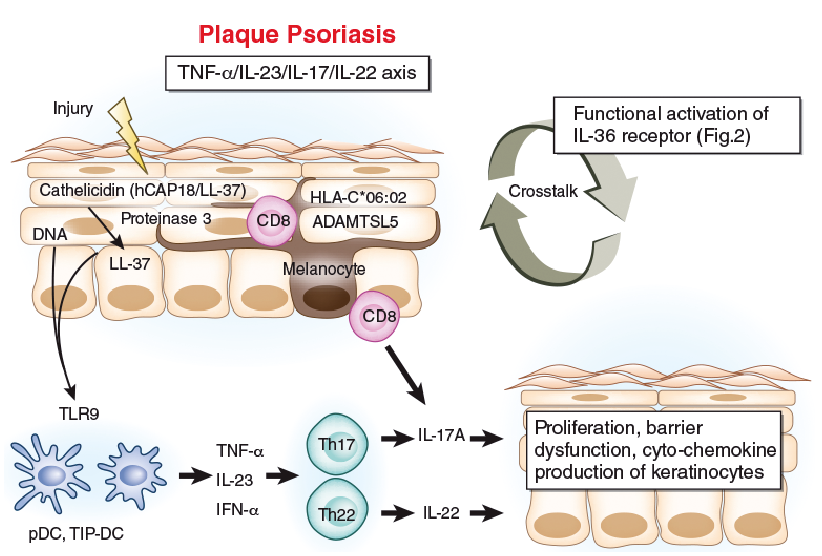
Plaque psoriasis occurs preferentially on areas susceptible to frequent friction or minor trauma, as represented by Koebner’s phenomenon (55–58). Cathelicidin (LL-37) is one of the anti-microbial peptides produced by keratinocytes and neutrophils (59). Cutaneous trauma, such as incisional injury, induces the expression of functional cathelicidin at the injured site (59). Human cathelicidin (hCAP18/LL-37) is encoded by the CAMP gene, and active LL-37 is released from pro-form hCAP18 via limited proteolysis exerted by proteinase 3 (Fig. 1) (60). An initial trigger of psoriasis is believed to be the activation of plasmacytoid DCs being stimulated by complexes of host DNA and LL-37, which are produced by keratinocytes after minor injuries (37, 38, 61, 62) (Fig. 1). Activated plasmacytoid DCs and damaged keratinocytes produce interferon (IFN)-α, IFN-β and TNF-α, which result in further production of TNF-α and IL-23 by plasmacytoid and recruited inflammatory DCs (TNF/iNOS-producing DCs or TNF-α/inducible nitric oxide synthase, producing dendritic cells [TIP-DCs]) (38, 62–65). IL-23 is critically involved in the generation and activation of IL-17- and IL-22-producing effector Th (Th17 and Th22) cells (38, 66, 67). Th17 cells have been detected in the blood and lesional skin, and they readily produce IL-17A and IL-22 (67, 68). In addition, the pivotal role of intraepidermal CD8+ T cells has been documented in psoriasis (69–71). These intraepidermal CD8+ T cells produce pathogenic IL-17A, and neutralization of CD8+ T cells effectively prevented psoriasis development in xenotransplanted mice model in vivo (71). Moreover, a recent study by Arakawa et al. (72) showed that intraepidermal CD8+ T cells recognize ADAMTS-like protein 5 (ADAMTSL5) on melanocytes, in concert with HLA-C*06:02, which is the main psoriasis risk allele.
Fig. 1. Pathogenesis of plaque psoriasis. Psoriatic lesions are frequently triggered by minor trauma (Koebner’s phenomenon). Skin injury causes dead keratinocytes to release self-DNA and cathelicidin (LL-37). Full-length cathelicidin (hCAP18/LL-37) is cleaved into active form LL-37 by proteinase 3. Self-DNA and LL-37 complex activate Toll-like receptor 9 (TLR9) in plasmacytoid dendritic cells (pDCs) and tumour necrosis factor (TNF)-α/inducible nitric oxide synthase, producing dendritic cells (TIP-DCs) to produce interferon-α (IFN-α), TNF-α and interleukin (IL)-23. IL-23 facilitates Th17 and Th22 cell proliferation. IL-17A and IL-22 induces the proliferation, barrier dysfunction and diverse cyto-chemokine production of epidermal keratinocytes, leading to epidermal acanthosis, parakeratosis and inflammatory cell infiltration, including neutrophils. The lesional epidermis harbours CD8+ T cells which target ADAMTS-like protein 5 (ADAMTSL5) on melanocytes and produce IL-17A. The “TNF-α/IL-23/IL-17/IL-22 axis” and the “functional activation of IL-36 receptor” (Fig. 2) are closely related to each other.
IL-17A upregulates the proliferation of keratinocytes and downregulates their differentiation (73). IL-22 also drives epidermal hyperplasia, primarily by the downregulation of genes involved in terminal differentiation (74, 75). IL-17A acts on keratinocytes to induce chemokines that lead to neutrophil, TIP-DC, and Th17 cell influx into the skin (76). TIP-DCs produce inducible NO synthase (leading to NO production, which can dilate dermal blood vessels) and secrete TNF-α, IL-20 and IL-23 (63, 77, 78). IL-17A upregulates the production of the neutrophil-attractive chemokines CXCL1, CXCL2 and CXCL8 by keratinocytes (4, 79). IL-17A also promotes the expression of TNF-α by keratinocytes (80), indicating that the aforementioned TNF-α/IL-23/IL-17/IL-22 axis probably forms a vicious cycle in the development of psoriasis lesions (Fig. 1). The mechanistic hypothesis coincides with the remarkable clinical effects of the antibodies against TNF-α, IL-23, IL-17A on both psoriasis and pustular psoriasis (81–83), further emphasizing the canonical role of the TNF-α/IL-23/IL-17/IL-22 axis in psoriatic pathogenesis.
In addition to the TNF-α/IL-23/IL-17/IL-22 axis, the gene expression levels of IL-36α, IL-36β, IL-36γ and IL-36Ra are all significantly elevated in lesional psoriatic skin, compared with normal control skin. However, only IL-36γ and IL-36RN exhibited hierarchical increases in the ascending order of normal < non-lesional psoriatic < lesional psoriatic skin (41, 84, 85). Moreover, treatment using etanercept, a human TNF receptor fusion protein, significantly decreased the lesional gene expression levels of IL-36α, IL-36γ and IL-36Ra (41). Carrier et al. (86) reported that the gene expression levels of IL-36α, IL-36β and IL-36γ were all elevated in the lesional skin of psoriasis and that this increase was strongly correlated with the expression levels of IL-22, IL-17A and TNF-α, respectively, but not of IL-17F. In addition, elevated IFN-γ expression was detected in lesions and was correlated well with the expression of all IL-36 members (86). On immunohistology, upregulated expression of IL-36γ was detected in the increasing order of lesional psoriatic > non-lesional psoriatic > normal control skin (49, 85).
Boutet et al. (39) compared the expression of IL-36 members and IL-36Ra in psoriasis, rheumatoid arthritis and Crohn’s disease in humans and in murine models. In an imiquimod-induced murine psoriatic model and in human psoriasis, the expression levels of IL-36α, IL-36γ and IL-36Ra, but not IL-36β, were correlated with IL-1β and Th17-related cytokines (IL-17A, IL-22, IL-23, and CCL20). In mice with collagen-induced arthritis and in the synovium of patients with rheumatoid arthritis, IL-36α, IL-36β, IL-36γ and IL-36Ra were all elevated and were correlated with levels of IL-1β, CCL3, CCL4 and macrophage colony-stimulating factor, but not with Th17-related cytokines. In the colons of mice with dextran sulphate sodium-induced colitis and in patients with Crohn’s disease, only IL-36α and IL-36γ were induced at low levels and were correlated with IL-1β and IL-17A. Overall, only a minor subgroup of patients with rheumatoid arthritis (17–29%) or Crohn’s disease (25%) has an elevated IL-36 agonist/receptor antagonist ratio, which is in sharp contrast to its elevation in as many as 93% of patients with plaque psoriasis (39).
These results suggest that IL-36 members are indeed overexpressed in the lesional skin of plaque psoriasis and that the “functional activation of IL-36R” might contribute to the persistence and perpetuation of psoriatic inflammation, together with the TNF-α/IL-23/IL-17/IL-22 axis (Fig. 1). To support this notion, the effectiveness of anti-IL-36R blocking antibody was confirmed in a human psoriatic skin xenotransplantation model (78). SCID mice transplanted with human psoriatic lesional skin were treated with an anti-human IL-36R antibody that blocked the actions of IL-36α, IL-36β and IL-36γ. Anti-IL-36R antibody substantially reduced the epidermal hyperplasia and other skin changes associated with psoriasis as effectively as etanercept (78). The anti-human IL-36R antibody did not cross-react with murine IL-36R, and murine IL-36α, IL-36β and IL-36γ did not signal through human IL-36R (78).
A different treatment response to biologics indicates that the pathogenesis of plaque psoriasis and pustular psoriasis does not completely overlap (4). The “functional activation of IL-36R” is likely to be more closely associated with pathogenesis in generalized pustular psoriasis, pustule formation of plaque psoriasis and palmoplantar pustulosis than with plaque psoriasis (Fig. 2) (4, 5, 54). The transcriptome of generalized pustular psoriasis shares features of plaque psoriasis, but is skewed toward innate immune inflammation, including IL-1 and IL-36 (4, 5). The expression levels of IL-1β, IL-1RN, IL-36α, IL-36β, IL-36γ and IL-36Ra are significantly increased, both in generalized pustular psoriasis and plaque psoriasis, compared with normal skin, with significantly greater expressions of IL-1β, IL-36α and IL-36γ in generalized pustular psoriasis than in plaque psoriasis (4, 5). In contrast, the expression levels of Th17/Th1-related cytokines, such as IL-17A, IL-22, IL-23p19, IFN-γ and IL-18, are significantly increased in plaque psoriasis compared with the levels observed in generalized pustular psoriasis and in normal skin (4).
Fig. 2. Pathogenesis of pustular psoriasis. Skin injury causes dead keratinocytes to release double-stranded RNA (dsRNA) and cathelicidin (LL-37). LL-37 or dsRNA (possibly by TLR3 ligation) activates surrounding keratinocytes to produce interleukin (IL)-36 members. LL-37 and dsRNA complex activate mitochondrial antiviral-signalling protein (MAVS) in the surrounding keratinocytes to release interferon (IFN)-β. The ligation of IL-36R by IL-36 enhances the production of diverse chemokines and assembles neutrophils, T cells, dendritic cells (DCs) and monocytes. Full-length IL-36 is processed and activated by neutrophilic proteases, as well as keratinocyte-derived cathepsin S. The IL-36 receptor antagonist (IL-36Ra/IL36RN) inhibits IL-36R activation by competitive inhibition. The functional activation of IL-36R is preferentially involved in the pathogenesis of pustular psoriasis, and conversely, the TNF-α/IL-23/IL-17/IL-22 axis is driven more efficiently in plaque psoriasis (Fig. 1). However, these 2 pathways are closely related and cross-talk with each other, forming a positive inflammatory loop.
In parallel with the augmented IL-1and IL-36 signals in generalized pustular psoriasis compared with plaque psoriasis, the expression levels of neutrophilic chemoattractants, CXCL1, CXCL2 and CXCL8, are also significantly and more abundantly increased in generalized pustular psoriasis than in plaque psoriasis (4). Infiltrated neutrophils were positively stained with cathepsin G, neutrophil elastase and proteinase 3 in the lesional skin of psoriasis and pustular psoriasis. Notably, IL-36α was processed and activated by neutrophil elastase, but not by cathepsin G, and its processing was abrogated by serpin A1, a specific inhibitor of neutrophil elastase. In contrast, IL-36γ was processed and activated by cathepsin G, but not by neutrophil elastase, and its processing was prevented by serpin A3, a specific inhibitor of cathepsin G. Activated IL-36α and IL-36γ were capable of enhancing the expression levels of CXCL1, CXCL2, CXCL8 and CCL20 from keratinocytes (4). Moreover, IL-17A induced IL-36 more potently in human psoriasis-derived keratinocytes than in healthy keratinocytes, while IL36RN expression remained unaffected (87).
On immunohistology, the expression of IL-36γ was localized in the suprabasal to granular layer of the epidermis (especially peripustular keratinocytes), and staining of the basal layer was minimal in generalized pustular psoriasis and plaque psoriasis (4, 49, 85). The immunohistological expression of IL-36α was also confirmed in the upper spinous layer to granular layer in generalized pustular psoriasis and plaque psoriasis, especially in peripustular keratinocytes (4). Using an in situ hybridization technique, the expression of IL-36α was also strongly detected in the superficial layers of the epidermis in the lesional skin of generalized pustular psoriasis and plaque psoriasis, but not in chronic eczema or nummular eczema (88).
In contrast to the high proportion (46–82%) of IL36RN mutant carriage in generalized pustular psoriasis (42, 53), it was lower (10–17%) in pustular cases of plaque psoriasis (54). Transcriptome assay also demonstrated the importance of IL1A/B, IL36A/B/G/RN and the neutrophil chemokines CXCL1/CXCL8 in palmoplantar pustulosis (5). However, the BBS1 and TWIST1 genes related to limb malformation are specifically linked to palmoplantar pustulosis, but not to generalized pustular psoriasis (5). In addition, STEAP1 and STEAP4, expressed in keratinocytes, are key players for regulating IL1A/B, IL36A/B/G/RN and CXCL1/CXCL8 (5).
These results emphasize the critical roles of IL-36/(IL-1β) in the pathogenesis of pustular psoriasis. However, it should be borne in mind that Th17/(Th1) signalling is also an active and common player in both pustular psoriasis and plaque psoriasis (4, 5) (Figs 1 and 2).
Murine models have shown the diverse action of IL-36 agonists on keratinocytes and immunocytes. Transgenic overexpression of IL-36α (K14/Il36a transgenic) in basal keratinocytes in mice resulted in epidermal thickening, hyperkeratosis, mixed inflammatory cell infiltrate (CD205+DCs, CD3+ T cells, macrophages and neutrophils) and elevated chemokine expression (CCL2, CXCL6, CCL7, and CXCL2), manifesting in newborns, while the phenotype is not dependent on mature T cells because it is not cancelled in T-cell deficient Rag2–/– mice (88). Backcrossing to an Il36rn-deficient mouse augments the IL-36α transgenic phenotype, reflecting the effects of nonsense IL36RN mutations in human pustular psoriasis (88). The IL-36α transgenic phenotype is partly cancelled by the neutrophil-depleting antibody or TNF-α-neutralizing antibody, suggesting roles for both neutrophils and TNF-α in IL-36α-induced skin changes (88).
Interestingly, the epidermal and dermal changes in K14/Il36a transgenic mice resolve by 3 weeks of age, but reappear at approximately 6 months of age (88). However, the epicutaneous application of 12-O-tetradecanoylphorbol-13-acetate (TPA) to the resolved and phenotypically normal skin of K14/Il36a transgenic mice elicited severe skin inflammation, similar to human psoriasis (78). The TPA-treated transgenic skin was reddened, thickened, scaly and crusted with acanthosis, hyperkeratosis, parakeratosis, hypogranulosis and neutrophilic microabscesses in the spinous and cornified layers, a mixed dermal infiltrate containing macrophages/DCs, neutrophils and lymphocytes, and an increase in and dilation of superficial dermal blood vessels (78). Like human psoriasis, IL-17A, IL-22, IL-23 (both p19 and p40 subunits), and the antimicrobial peptides S100A8, S100A9 and β-defensin 4 (an orthologue of human β defensin 2) were significantly upregulated in TPA-treated transgenic skin. IL-1β, a regulator of Th17 differentiation, was also upregulated (78). Notably, psoriasiform eruption in TPA-treated transgenic mice showed significant improvement by injection of anti-TNF-α, anti-IL-12/23p40 or anti-IL-23p19 antibody (78). Intradermal injection of IL-36α into wild-type FVB mice led to substantial increases in IL-36α, IL-17A, IL-23, TNF-α and IFN-γ mRNA. Conversely, intradermal injections of IL-17A, IL-23, TNF-α, IFN-γ, IL-22 or their combination upregulated the mRNA expression of IL-36α, as well as IL-36β and IL-36γ (78).
The skin changes in the K14/Il36a transgenic mice were distinct from those generated in K14/ Il1a and K5/Il18 transgenic mice. The K14/Il1a transgenic mice exhibited marked hair loss without any prominent epidermal acanthosis. The dermal infiltrates were mainly macrophages in the K14/Il1a transgenic mice (89). The K14/Il36a transgenic mice had increases in epidermal thickness and in the numbers of neutrophils, CD205+ DCs and T cells (88, 89). In contrast, the K5/Il18 transgenic mice did not have overt macroscopic or microscopic abnormalities in the skin (90). However, the contact hypersensitivity reaction to trinitrochlorobenzene in the K5/Il18 transgenic mice was significantly intensified compared with wild-type counterparts, resembling atopic dermatitis featuring acanthosis; an infiltrate composed of eosinophils, neutrophils, and mast cells; as well as an increase in serum IgE levels (90).
Intradermal injection of murine IL-36α into CD1 mice every other day for 10 days induced mild epidermal acanthosis and pronounced leukocytic infiltrate without any visible skin changes (50). The infiltrated dermal cells were largely CD11b+ neutrophils with few CD4+CD8– T cells. The gene expression of the injected skin revealed upregulation of Ccl3, Ccl4, Cxcl12, Il1b and Hbegf (50).
Imiquimod (TLR7 agonist)-induced skin inflammation is currently well investigated as a murine model for psoriatic inflammation (41, 91). Topical treatment with imiquimod of skin cancer induces psoriasis in susceptible patients (92, 93). The topical application of imiquimod on the ears of C57BL/6 mice induced ear swelling, erythema, skin flaking, acanthosis and hyperkeratosis with dermal infiltration of IL-17A+ γδ T cells, neutrophils, macrophages and IL-23+ DCs (91). These epidermal and dermal features were strongly inhibited in mice deficient in Il36r and were augmented in mice lacking Il36rn (91). Moreover, imiquimod-induced ear thickness was modestly protected against in il17a–/– and il22–/– mice and was slightly more potently inhibited in il23a–/– mice, probably because IL-23 can induce both IL-17- and IL-22-producing cells. Consistent with this notion, ear thickness and neutrophil infiltrates were further ameliorated in il17a–/– mice when combined with injection of anti-IL-22 neutralizing antibody (91). The IL-23-mediated expansion of IL-17A-producing γδ T cells was reported to depend on IL-1 (94). Nevertheless, IL-1R1 deficiency did not protect mice from imiquimod-induced psoriasiform dermatitis. Ear swelling, acanthosis, and skin flaking were comparable in Il1r1–/– and wild-type mice. Only dermal neutrophilia was suppressed in Il1r1–/– mice (91). Among these knockout mice, imiquimod-induced psoriasiform inflammation, including epidermal thickness and dermal neutrophilic infiltration, was most inhibited in Il36r–/– mice because: (i) IL-36 induces the production of CXCL1 and CCL20 from keratinocytes and fibroblasts and attracts neutrophils and T cells; (ii) IL-36 upregulates the expression of the keratinocyte mitogens, such as granulocyte colony-stimulating factor and transforming growth factor α in keratinocytes; and (iii) IL-36 induces the production of IL-36 in keratinocytes in an autocrine fashion (91). Because keratinocytes are devoid of TLR7, imiquimod initiates IL-36 production by activating TLR7+ CD11c+ DCs. The released IL-36 upregulates the production of IL-23 from activated DCs, as well as the proliferation and chemokine induction of keratinocytes. The recruited T cells are directed by IL-23 to produce IL-17A and IL-22, and these Th17 cytokines further accelerate keratinocyte proliferation and neutrophilic infiltration (91). Although the aforementioned murine models were useful in investigating psoriatic inflammation, no animal models exist so far for pustular psoriasis.
As demonstrated in the above murine studies in vivo, the IL-36 agonists interact with LL-37, IL-17, IL-22 and IFN to enhance the inflammatory response in human keratinocytes in vitro, and the cross-talk among these psoriatic factors is important to manifesting clinical psoriasis. In addition to skin injury (55), the administration of type I IFN (IFN-α and IFN-β) exacerbates psoriasis (95). The role of IFN-α or IFN-β in psoriasis has also been reported in mice lacking the type 1 IFN receptor or in mice treated with IFN-α- or IFN-β-neutralizing antibodies because both systems failed to develop T-cell-mediated skin inflammation (61, 96). LL-37 links skin injury to type I IFN production in human keratinocytes (59, 61, 64, 96). LL-37, together with double-stranded RNA (dsRNA) released from damaged keratinocytes, potently upregulates the production of IFN-β from keratinocytes, indeed promoting the activation and maturation of DCs (64). Human keratinocytes express the highest amounts of mitochondrial antiviral-signalling protein (MAVS) mRNA and moderate mRNA levels of TLR3, while other TLRs, including TLR4, TLR7, TLR8, and TLR9, are barely detectable (64). Poly [I:C] (dsRNA) activates TLR3, but not MAVS and causes keratinocytes to produce IFN-β, while LL-37 and dsRNA complex activate both TLR3 and MAVS and induce higher production of IFN-β (64). Notably, CRAMP, the murine homologue of LL-37, fails to enhance poly [I:C]-induced IFN-β expression, suggesting that the synergistic effect is specific to human LL-37 (64) (Fig. 2). In contrast, plasmacytoid DCs express high levels of TLR7 and TLR9 (64). LL-37 binds to self-DNA, and the LL-37/self-DNA complex induces IFN-α expression in plasmacytoid DCs through TLR9 (Fig. 1) (62).
In the lesional skin of psoriasis, overexpressed LL-37 co-localized with IFN-β expression (64). Notably, upregulated expression of IL-36γ coexists with LL-37 expression in psoriatic lesions (49). LL-37 significantly increases functional IL-36γ expression in both undifferentiated (low calcium) and differentiated (high calcium) keratinocytes, whereas the enhancing effect is stronger in the differentiated condition (49). The IL-36γ-inducible effect of LL-37 is dependent on the Gi protein-coupled receptor and p38 MAPK signalling (49). LL-37 upregulates the production of CXCL1, CXCL8, CXCL10 and CCL20 from human keratinocytes, in part via released IL-36γ (49). In accordance with this notion, LL-37 also enhances IL-36R and IL1RAP (49). These studies emphasize the importance of LL-37 as an injury sensor in the epidermis.
dsRNA could also directly enhance the gene and protein expression levels of IL-36β and IL-36γ in human keratinocytes (47). Human keratinocytes were challenged with a panel of TLR ligands, including poly [I:C] (TLR3 ligand), flagellin (TLR5 ligand), E. coli DNA (TLR9 ligand), lipoteichoic acid (TLR2 ligand), lipopolysaccharide (TLR4 ligand) and zymosan (TLR2 ligand) (47). The gene expression levels of IL-1α and IL-1β were upregulated by these ligands at 1, 6 and 24 h, while the robust upregulation of IL-36β and IL-36γ was induced only by poly [I:C] and flagellin at 6 and 24 h after stimulation. Lipopolysaccharide and zymosan weakly upregulated IL-36γ gene expression at 24 h (47). The release of IL-36γ protein was observed only by poly [I:C] but not by flagellin. The extracellular release of IL-36γ was augmented in the presence of 5 mM ATP. The secretion of IL-36β protein was not detected by any stimulation. Poly [I:C], but not flagellin, caused cell death (pyroptosis) of keratinocytes in a caspase-3/7-dependent manner, and caspase-3/7 is a prerequisite for the release of IL-36γ (47). In addition, caspase 1 activation is required for IL-36γ gene expression and caspase 3/7 activation (47). These results also suggest the importance of IL-36γ as an injury sensor in the epidermis.
Both IL-17 and IL-22 and TNF-α are potent inducers of IL-36α, IL-36β and IL-36γ in human keratinocytes (86) (Fig. 1). IL-22, in concert with IL-17 or TNF-α, synergistically or additively enhances the production of IL-36α, IL-36β and IL-36γ from keratinocytes. Although IL-17A and TNF-α induce all 3 IL-36 members in keratinocytes, IL-17A most strongly induces the expression of IL-36α, and TNF-α strongly induces IL-36β. Moreover, the expression levels of IL-36α, IL-36β and IL-36γ are induced by either IL-36, especially in the simultaneous presence of IL-17A, or by TNF-α (86). IL-36α, IL-36β and IL-36γ also upregulate the expression levels of TNF-α, IL-6 and IL-8, respectively, which are further augmented by the simultaneous addition of IL-17A or TNF-α. IFN-γ selectively activates the expression of IL-36β, but not IL-36α or IL-36γ (86). Johnston et al. (41) reported similar findings that the gene expression of IL-36γ in human keratinocytes was augmented by each of the psoriasis-associated cytokines, i.e. TNF-α, IL-1α and IL-17A. Although IL-22 alone is ineffective to induce IL-36γ gene expression, it synergistically potentiates the upregulatory effects of IL-17A. The synergistic action of IL-36γ induction is evident in combination with IL-1α+ TNF-α or IL-1α+IFN-γ (41). These in vitro studies using skin-resident keratinocytes indicated the active and mutual cross-talk of the TNF-α/IL-23/IL-17/IL-22 axis and the functional activation of IL-36R.
Psoriasis is considered a human-specific disorder. Unlike human keratinocytes, murine cathelicidin CRAMP fails to activate MAVS in combination with dsRNA (64). Murine keratinocytes do not produce IL-1β (97, 98). The characteristics of human keratinocytes might explain the human-specific enigma. Plaque psoriasis and pustular psoriasis have relevant and partially overlapping, but distinct, clinical features. The TNF-α/IL-23/IL-17/IL-22 axis is the common pathogenetic pathway in these disorders, because blockade of IL-23 by ustekinumab is effective in generalized pustular psoriasis regardless of IL36RN mutation status (99). Nevertheless, the functional activation of IL-36R by agonists and the loss of function mutation of its antagonist IL-36Ra are implicated to be key pieces in a pathogenetic jigsaw puzzle. Considering: (i) the potent proliferative activity of IL-17 and IL-22 on human keratinocytes; (ii) the lack of clubbed regular acanthosis in pustular psoriasis; and (iii) the effective chemoattraction of neutrophil by IL-36 agonists; functional IL-36R activation is preferentially involved in the pathogenesis of pustular psoriasis, and conversely, the TNF-α/IL-23/IL-17/IL-22 axis more efficiently drives plaque psoriasis (Figs 1 and 2). Notably, these 2 pathways are closely related and cross-talk with each other, forming a positive inflammatory loop. Although the agonistic activities of IL-36α, IL-36β and IL-36γ on IL-36R are redundant, current studies have supported the importance of IL-36γ (and IL-36α) in human psoriatic inflammation (4, 39, 41, 48, 49, 85, 88). The biological significance of each isoform of IL-36R agonists warrants future investigation.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize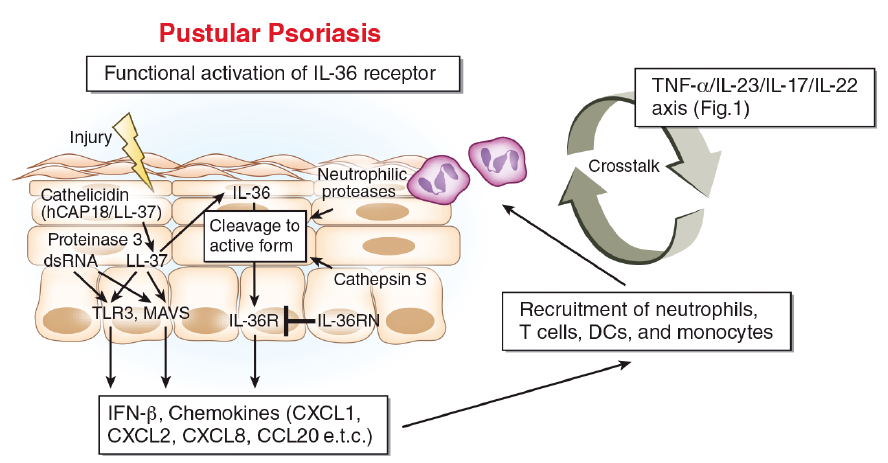 Click to show fullsize
Click to show fullsize