


1Department of Dermatology and Venereology, 3Department of Otorhinolaryngology and 4Department of Ophthalmology, Medical Center, Faculty of Medicine, University of Freiburg, and 2Berta-Ottenstein-Programme, Faculty of Medicine, University of Freiburg, Freiburg, Germany. E-mail: cristina.has@uniklinik-freiburg.de
Accepted Mar 27, 2018; Epub ahead of print Mar 27, 2018
Junctional epidermolysis bullosa (JEB) is a hereditary blistering disease caused by reduced dermal–epidermal adhesion due to deficiencies in laminin 332, collagen XVII or integrin α6β4 (1). In JEB caused by laminin 332 deficiency, chronic wounds appearing in infancy can be extremely resistant to therapy, and may determine a severe course (2) and lethal outcome (3). We report here a successful multidisciplinary approach used to treat severe chronic hypergranulating facial wounds and scarring in 2 siblings with JEB generalized-intermediate due to a LAMB3 splice site mutation.
The children were born to healthy consanguineous parents from Libya.
The girl (case 1) first presented to our centre at the age of 9 years 2 months. Mild skin blistering had started at birth, and her nails were missing. After the age of 8 years, she developed chronic wounds on her neck and nose, leading to progressively blocked nostrils (Fig. 1a). Oral blistering was reported, and her teeth showed mild enamel defects. Her weight (23.7 kg) was in the low normal range. She had microcytic anaemia (haemoglobin 5.5 g/l, normal 12–16 g/dl) and vitamin D deficiency (4.4 ng/ml, normal 20–70 ng/ml).
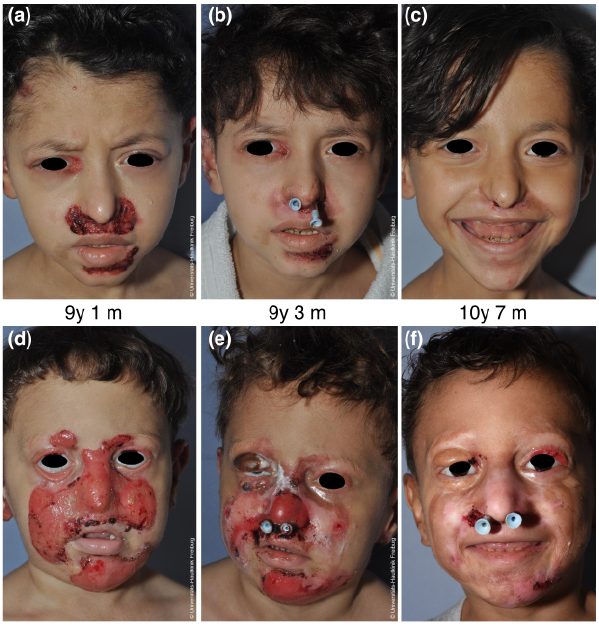
Fig. 1. Case 1 (upper panel) and case 2 (lower panel) with chronic facial wounds in junctional epidermolysis bullosa generalized-intermediate. At initial presentation, mid-facial wounds, crusts and scarring were present in both siblings. (a, d) Note the completely obstructed nostril and the ectropion in the boy (d). During the course of treatment, wound size decreased and crusts reduced. (b, e) At last presentation, mostly post-inflammatory hyper- and hypo-pigmentation and few crusts were present. (c, f) Nasal placeholders were inserted to relieve nasal obstruction; they were removed after 1 month in the girl with a good cosmetic and functional result (c). In the boy, nasal tubes were replaced twice and are currently still in place (f). The boy’s ectropion on the right eyelid was corrected using an autogenous skin graft from the upper arm for the upper eyelid and tarsorraphy was performed for the lower eyelid (e; 3 weeks after surgery). After postoperative swelling had resolved, the functional result is good and the cosmetic result satisfactory, but ectropion on the left eye shows progress (f). Age at photography is indicated in years (y) and months (m). Written permissions are given by the parents to publish these photos.
Her brother (case 2) was first seen at the age of 4 years 5 months. Blistering had also started at birth, nails were missing and he had amelogenesis imperfecta. The boy had had severe facial wounds since infancy. His nostrils were blocked by scarring, so that only oral breathing was possible and his sleep was disturbed. Shrinkage of the eyelid tissue had led to ectropion of both upper and lower lids with consecutive lagophthalmos and exposure keratoconjunctivitis (Fig. 1d). The boy experienced feeding difficulties due to obligate oral breathing and was underweight (14.2 kg, just above the 3rd percentile (4)) with severe microcytic anaemia (haemoglobin 4.8 g/l).
Wound swabs revealed Streptococcus pyogenes (Group A), and methicillin-resistant Staphylococcus aureus (MRSA) was isolated repeatedly. Fungal superinfections and leishmaniosis were ruled out.
Immunofluorescence mapping (5) showed junctional skin cleavage with almost complete depletion of laminin 332. Mutation analysis identified the homozygous splice-site mutation c.2701+1G>A in exon 18 of LAMB3. Both parents were heterozygous for c.2701+1G>A.
When the siblings had just moved to Germany, wound care was delivered by the parents, but was mainly refused by the boy. Olive oil was applied for skin care. Fusidic acid alone, or with betamethasone (Fucidine®/Fucicort®) had been used for more than one year up to 3 times daily with no effect; discontinuation lead to worsening of the wounds. Systemic antibiotics had been administered intermittently. Surgical interventions were avoided because of the severe inflammation and scarring around the designated operation sites.
Topical treatment with antiseptics (polyhexanide), steroids (prednicarbate, halometasone/triclosan cream (Infectocortisept®)), as well as panthenol ointment, 1% coriander oil in cold cream and soft zinc paste, was initiated. Streptococcus pyogenes infections were treated with oral penicillin V and MRSA colonisation with a standard regime.
After 8 weeks of topical treatment, when inflammation and granulation were markedly reduced (Fig. 1b and e), both children received otorhinolaryngological interventions in general anaesthesia. Scar tissue was excised, nasal stenosis dilated and the nostrils were reopened in the boy. Plastic tubes (aspirator hoses size 10) were inserted as placeholders via the nasopharynx and fixated by breathing hose adapters. In the girl, these tubes were removed after 6 weeks; in the boy, the tubes were exchanged after 3, 7 and 16 months and are currently still in place.
The boy’s ectropion of the right upper eyelid was also corrected after 8 weeks of topical treatment. Subciliary incisions were followed by separating vertical tractions. The skin defect was covered by an autogenous skin graft from the medial upper arm. To prevent traction formation during wound healing, a temporary tarsorrhaphy was performed for 2 weeks (Fig. 1e). After the postoperative swelling had resolved, the functional and cosmetic results were very satisfactory (Fig. 1c and f).
The children in our study had disfiguring mid-facial wounds with severe scarring, compromising the functionality of the anatomical structures. The LAMB3 mutation found in the siblings was recently reported in a Libyan patient with JEB (6). The mutation was predicted to abolish the donor splice site and lead to an aberrant transcript with an in-frame skipping of 82 nucleotides, but the precise consequences have not been disclosed (6). The current cases demonstrate that residual laminin 332 expression accounts for a generalized-intermediate JEB phenotype.
Granulation tissue is a common feature of JEB (1, 7), yet its treatment remains difficult. A case of JEB caused by a homozygous LAMB3 mutation with a hypergranulating facial wound was reported recently, but with no effective therapy (8). In our patients, severe complications such as corneal scarring or ulcers with permanent loss of vision and impaired breathing, were imminent, necessitating a coordinated multidisciplinary approach. We decided to attempt surgical intervention following topical treatment, as a conservative approach could not have addressed nasal stenosis and ectropion.
While nasal crusting has been reported in EB (9), complete nasal stenosis due to scarring as seen in case 2 (Fig. 1f) has not. We show that, after reduction of facial wounds through topical treatment, surgical intervention for nasal stenosis is possible and successful. Both children experienced symptom relief and increased quality of life as a result of both procedures. The girl has no evidence of recurrent nasal stenosis and the chronic wounds in the periocular region healed with topical therapy. Nasal placeholders are still in situ in the boy at 21 months post-intervention, and eyelid function was fully restored by surgery following local therapy.
In addition to their chronic wounds, the children had microcytic anaemia, a common feature of generalized JEB associated with chronic wounds and inflammation (3). Anaemia in JEB may negatively affect wound healing and vice versa. A case of JEB generalized-intermediate in which hypergranulating wounds led to severe anaemia was successfully treated with colchicine (10).
In the two cases reported here, the chronic facial wounds vastly improved on long-term topical therapy with triclosan/halomethasone, supporting the guideline recommendations (11). Next to the improvement in wounds, anaemia stabilized during the observation period, suggesting that potent topical steroids are justified in JEB and can pave the way for surgical intervention to prevent complications.
The technical support by Ioannis Athanasiou and Kaethe Thoma is gratefully acknowledged. The authors thank Professor J. Kohlhase for sequencing the DNA samples.
Funding sources. AR is supported by the Berta-Ottenstein-Programme for Clinician Scientists, Faculty of Medicine, University of Freiburg.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize