


1Department of Dermatology, Xinhua Hospital, Shanghai Jiaotong University School of Medicine, 1665, Kongjiang Road, Shanghai 200092, and 2Center for Rare Disease, Shanghai. *E-mail: liming01@xinhuamed.com; zryaosmu@sohu.com
#These authors contributed equally to this paper.
Accepted Sep 4, 2018; Epub ahead of print Sep 5, 2018
Ultraviolet-sensitive syndrome (UVSS, OMIM 600630,614621,614640) is a rare autosomal recessive genodermatosis, characterized by isolated cutaneous photosensitivity and deficiency in transcription-coupled repair (TCR), a subpathway of nucleotide excision repair that rapidly removes transcription-blocking DNA damage (1). UVSS was first described by Itoh et al. (2). Affected individuals are sensitive to the ultraviolet rays in sunlight; a small amount of sun exposure can induce acute sunburn, dryness, freckling, pigmentation anomalies, and telangiectasias in sun-exposed areas of the skin. Although exposure to UV can cause skin cancers, patients with UV-sensitive syndrome do not have the pre-disposition to skin malignancy as in xeroderma pigmentosum. UVSS comprises 3 groups, UVSS-1, UVSS-2 and UVSS-3, caused by mutations in ERCC8 (CSA), ERCC6 (CSB) and UVSSA (KIAA1530), respectively (3). In 1995, Henning et al. (4) mapped the ERCC8 gene to chromosome 5p14-p12. In 2009, Nardo et al. (5) revealed that the pathological gene of UVSS-2 was the ERCC8 gene.
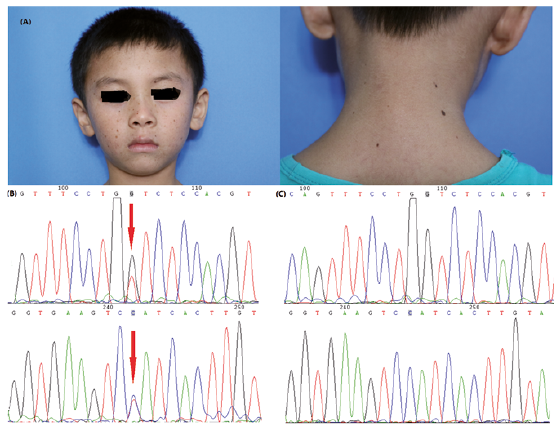
We report here a 6-year-old boy with a diagnosis of UVSS. He was the first child of healthy parents. The patient had had erythema and papules on his facial skin since birth. The neck and palmoplantar were involved and sporadic rice-sized hazel macules gradually appeared on his facial skin (Fig. 1). Sunburn makes symptoms worse, such as flush, desquamation, pruritus and so on, especially in sun-exposed areas. Medication appears to be ineffective. The patient had a slightly dark basal skin colour, lots of freckles, hypopigmented spots, telangiectasia, and slightly dried skin in sun-exposed areas, but no growth retardation or neurological abnormalities. He showed no pre-disposition to cutaneous tumours. Results of routine blood testing, trace elements examination, ANA, mycology examination, porphyrin and uroporphyrinogen were within normal ranges, except that intracellular zinc porphyrins were a little high. The result of the minimal erythema dose (MED) of normal skin to UV indicated that the patient is sensitive to UVB. (UVB 24.9 mJ/cm2, UVB < 30 mJ/cm2 signify the sensitivity to UVB).
Fig. 1. Ultraviolet-sensitive syndrome. (A) Clinical features of the patient, the sporadic rice-sized hazel macules on his face. (B) Compound heterozygous mutation c.582G>T, resulting in the mutation of p.W194C, and c.769G>A resulting in the mutation of p.G257R. (C) Part of the normal sequence from exon 7 and exon 9 of ERCC8 gene. Arrows indicate the mutations in this pedigree. Permission is obtained to publish these photos.
After obtaining informed consent, genomic DNA was extracted from the patient’s and his parents’ peripheral blood lymphocytes. The genomic DNA of this family was analysed using a gene panel, which consists of exons of 541 genes of genodermatosis. Mutations were identified by comparing with the reported cDNA reference sequence (GenBank accession number: NM_024666.3). In addition, our gene panel is comprised of the UVSSA and CSB genes. We did not detect any significant mutations in these (Table S1). This study was approved by the ethics committees of Shanghai Jiaotong University School of Medicine and conducted in accordance with the principles of the Declaration of Helsinki.
Sequencing results revealed 2 novel mutations. c.582G>T in exon 7 of ERCC8, originated from his mother, which results in the mutation of p.W194C and c.769G>A in exon 9 of ERCC8, originated from his father, which results in the mutation of p.G257R. Those 2 mutations segregated clearly with the disease phenotype within the family members, and c.582G>T was not detected in unrelated, healthy Chinese individuals. The variant frequency of c.769G>A in the ERCC8 gene is less than 0.01% in Chinese population.
The ERCC8 gene spans 53.7 kb and contains 12 exons. This gene encodes part of the CSA protein (DCX(ERCC8) complex), a DCX E3 ubiquitin-protein ligase complex containing ERCC8, RBX1, DDB1 and CUL4A, composed of 396 amino acid residues. The CSA complex interacts with RNA polymerase II; on UV irradiation it interacts with the COP9 signalosome and preferentially with the hyperphosphorylated form of RNA polymerase II (6). It also interacts with Cockayne syndrome type B (CSB) protein and with p44 protein, a subunit of the RNA polymerase II transcription factor IIH to activate the process of TCR and CSB was a substrate of this E3 ubiquitin-protein ligase. In addition, CSA belongs to the WD40-repeat family of proteins, which are highly conserved repeated units usually ending with Trp-Asp (WD). WD40-repeat proteins are found in all eukaryotes regulating a variety of cellular functions, such as cell-fate determination, gene transcription, mRNA modification, transmembrane signalling, and cell division. (7)
The ERCC8 gene is the pathogenic gene of Cockayne syndrome, type A and UVSS-2, but symptoms of UVSS-2 are much milder. UVSS cells are defective in TCR, but have normal global genomic repair (8). Nardo et al. (5) hypothesized that the mild phenotype in this patient was due to the lack of cellular sensitivity to oxidative stress. The ability to repair oxidative lesions may distinguish individuals with UVSS from those with CS.
To date, 8 patients have been characterized with UVSS; 6 are Japanese and 2 are white (3). In the current study, we identified 2 novel heterozygous base substitution, c.582G>T(p.W194C) and c.769G>A(p.G257R) of the ERCC8 gene, which belong to compound heterozygous mutations. Multiple software types (SIFT, Mutation Taster, LRT et al.) predicted these mutations as harmful mutations. These mutations affect the synthesis of CSA protein, which is involved in transcription-coupled nucleotide excision repair. The ubiquitination and subsequent proteasomal degradation of ERCC6 may be repressed. However, more details about UVSS remain to be elucidated.
In conclusion, this study reports the first UVSS-2 case diagnosed by genetic testing in China. This finding reinforces that ERCC8 is the pathogenic gene for UVSS-2 and expands the spectrum of the ERCC8 gene.
The authors thank all subjects for their ongoing participation in this study. This work was supported by a grant from the National Nature Science Foundation of China (81472867) and a grant from Shanghai Municipal Education Commission-Gaofeng Clinical Medcine Grant Support (20161417).


 Click to show fullsize
Click to show fullsize