


Department of Pathology, Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing 210042, China. *E-mail: ch76ch@163.com
#These authors contributed equally to this work.
Accepted Sep 18, 2018; Epub ahead of print Sep 18, 2018
Severe combined immunodeficiency (SCID) is characterized by absent or non-functional T and B cells. A series of different genetic variants are known to cause SCID. Mutations in recombinase activating genes 1 and 2 (RAG1 and RAG2) are the most common mutations of T-B-NK+ SCID (1). During the development of T and B cells, these genes are responsible for the rearrangements of the variable, diversity and joining segments of T- and B-cell receptors (2). The first case of SCID caused by mutation in RAG1 was reported in 1996 by Schwarz et al. (3). Mutations at RAG1 located within human 11p13 chromosome result in many kinds of primary immunodeficiency diseases, including SCID, Omenn syndrome (OS) and selective immunoglobulin A deficiency (SIgAD) (4). We described here a patient who was diagnosed with SCID with cutaneous lymphoproliferative diseases caused by novel mutations in RAG1.
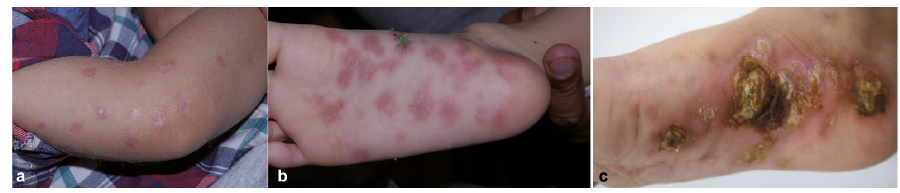
A 5-year-old boy was referred to our clinic with a 3-year history of asymptomatic infiltrated erythematous plaques on his trunk and limbs. He was otherwise healthy, except for frequent upper respiratory tract infections. His parents are not close relatives. Physical examination revealed multiple, asymptomatic, dull-coloured and irregular-shaped infiltrated erythematous plaques with a dry surface located on the trunk and limbs, and ulcerating erythemas or plaques with scab on his extremities (Fig. 1). Other physical examination was notable for mild hepatomegaly.
Fig. 1. Clinical pictures of the patient with severe combined immunodeficiency. (a, b) Dull-coloured, irregular-shaped infiltrated erythematous plaques located on the patient’s limbs. (c) Ulcers with scab on the patient’s feet.
Blood counts demonstrated lymphopaenia (1,008 cells/mm3). IgA level (0.06 g/l) was decreased significantly. A significant reduction in T and B lymphocytes was revealed by flow cytometry analysis (Table I). Routine laboratory tests, such as ANA, anti-dsDNA antibody, ENA and T-SPOT tests, were all normal. Epstein–Barr virus DNA was not detected in plasma. Bone marrow biopsy investigation was normal. A biopsy from the plantar skin showed hyperkeratosis and focal infiltration of lymphocytes with minimal atypia in the upper and middle dermis (Fig. S1). These cells were stained positively by CD4, CD5, CD20, CD56, CD68, CD79a, TIA-1 and GrB, but were negative for CD8, CD30 and CD31. The fraction of Ki-67 positive cells was 40%. The pathological diagnosis was lymphoproliferative disease (LPD).
Table I. Lymphocyte subsets of the patient
To investigate the genetic mutations in RAG1, we amplified DNA extracted from whole blood of the patient and family members who provided informed consent. Direct sequencing of the RAG1 amplification product identified novel compound heterozygous mutations in RAG1. Five heterozygous mutations were found at the cDNA positions of 813(c.813T>A), 870(c.870G>A), 2219(c.2219C>T), 2583(c.2583A>G) and 6440(c.6440G>A) in the patient. The patient’s father has 3 heterozygous mutations at cDNA positions, including 813(c.813T>A), 870(c.870G>A) and 6440(c.6440G>A) located in the RAG1. Sequence analysis also showed that the RAG1 of the patient’s mother was heterozygous for 2219(c.2219C>T) mutation and homozygous for 2583(c.2583A>G) and 6440(c.6440G>A) mutations. The RAG1 of the patient’s uterine sister was homozygous for 2583(c.2583A>G) and 6440(c.6440G>A) mutations (Fig. S2).
The patient was treated with a combination of prednisone and methotrexate and is preparing for hematopoietic stem cell transplant.
Primary immunodeficiency disease (PID) is a group of clinical syndromes characterized by the deficiency of immune organs, immune cells or immune reactive molecules caused by gene mutations. According to the update on the classification from the International Union of Immunological Societies Expert Committee in 2015, PID is divided into 9 categories, as follows: immunodeficiencies affecting cellular and humoral immunity; combined immunodeficiencies with associated or syndromic features; predominant antibody deficiencies; diseases of immune dysregulation; congenital defects of phagocyte number, function or both; defects in intrinsic and innate immunity; autoinflammatory disorders; complement deficiencies; and phenocopies of PID (5).
Compared with other PIDs, SCID is relatively common, with morbidity of 1/58,000–1/100,000 in children (6). Patients with SCID experience both severe defects in cellular immunity and humoral immunity. The patho-genesis of SCID is complicated, and a substantial majority of SCID are related to defect in the membrane and intracellular proteins. SCID is characterized by the development of the disorder. In rare cases, the dysfunc-tion of T, B and nature kill cells is involved. On the basis of defective lymphocyte subsets, SCID can be divided into 4 groups, namely T-B+NK+, T-B+NK-, T-B-NK+ and T-B-NK-. SCID can be caused by various defects or abnormalities of genes, such as IL2RG (19%), RAG1 (15%), IL7R (12%) and ADA (11%) (7).
The cutaneous symptoms of SCID mainly include eczematous lesions, erythroderma, non-infectious granuloma, microbial manifestations and LPD (8). LPD is present in more than 60 kinds of PID and commonly present in SCID, ataxia telangiectasia, Wiskott–Aldrich syndrome, X-linked lymphadenopathy, and Nijmegen breakage syndrome (9). LPD associated with PIDs may be expressed as reactive hyperplasia, pleomorphic lymphocytic hyperplasia or lymphomas (10). Slatter et al. (11) have described 2 cases of X-SCID manifested as LPD, which had CD20+ B lymphocyteinfiltration.
RAGs are located with the human chromosome 11p13 and consist of 2 adjacent genes, that is RAG1 and RAG2. DNA recombinant activating enzymes of RAG1 and RAG2 function are complex (12). Mutations in RAG1 cause primary immunodeficiency, including SCID, OS, and SIgA (4, 13). RAG1 mutation is the most common genetic defect leading to T-B-NK+ phenotype SCID, and the inheritance mode of SCID is autosomal recessive (1). Heterozygous compound mutations of RAG1 can also lead to PID. Sharapova et al. (14) presented a 14-year-old male with SCID caused by heterozygous compound mutations, i.e. c.256-257del and c.C1331T, in RAG1. Matthews et al. (15) reported a boy with OS bearing compound heterozygous mutations of RAG1.
We report here a case of SCID with LPD as the main clinical manifestation caused by the mutations of RAG1. His half-blooded sister has only 2 homozygous mutations including 2583(c.2583A>G) and 6440(c.6440G>A) mutations in the RAG1 and is healthy without SCID. We suppose that these 2 homozygous mutations do not cause SCID. His father has heterozygous mutations in 813(c.813T>AT) and 870(c.870G>AG) in RAG1 without SCID which suggests that these 2 mutations alone may not lead to SCID. Two possibilities can be attributed to this result; one is that the mutations of 813(c.813T>AT) and 6440(c.6440G>AG) together may cause SCID, and the other is that the mutations of 870(c.870G>AG) and 6440(c.6440G>AG) together may cause SCID.
To our knowledge, this work is the first SCID case with cutaneous LPD caused by novel compound heterozygous mutations in RAG1.


 Click to show fullsize
Click to show fullsize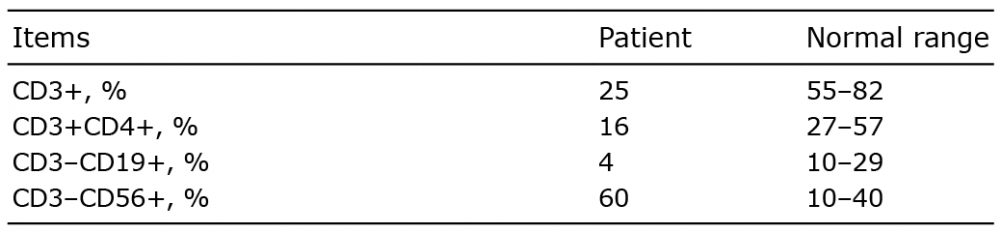 Click to show fullsize
Click to show fullsize