


Division of Dermatology, Department of Medicine of Sensory and Motor Organs, Faculty of Medicine, Tottori University, Yonago, Japan. E-mail: m0641023@med.tottori-u.ac.jp
Accepted Sep 18, 2018; Epub ahead of print Sep 18, 2018
Cytophagic histiocytic panniculitis (CHP) is a rare panniculitis that usually presents with liver failure and haemorrhage, leading to death in most cases (1). Histopathologically, skin lesions consist of benign histiocytes, including phagocytosed erythrocytes, leukocytes and platelets. We report here a rare case of CHP associated with myelodysplastic syndrome (MDS).
An 88-year-old man was admitted to our hospital with a 10-day history of high fever and painful plaques on his extremities. He had been on anti-angina medications for 40 years. Physical examination revealed a fever of 38.7°C and multiple erythematous, tender and indurated plaques on his arms and legs (Fig. 1). He had no evidence of lymphadenopathy. Blood analysis revealed an elevated total white blood cell count (10.3 × 103/μl; normal range 3.3–8.8 × 103) with elevated level of neutrophils (88%; 40–70%) and myelocytes (1.0%; 0%) and low level of haemoglobin (8.3; 12–17 g/dl). Platelet count was within the normal range (192 × 103; 125–343 × 103/μl). Chemistry profile results included elevations of alkaline transferase (AST) (41; 10–40 U/l), aspartate transaminase (AST) (49; normal range 13–30 U/l), C-reactive protein (18.9; normal range 0–0.15 mg/dl), ferritin (567; normal range 23.9–336.2 ng/ml) and α chain of soluble IL2 receptor (1,700; normal range 122–496 U/ml), prolonged activated partial thromboplastin time (40.1; normal range 23.3–38.2s), and normal prothrombin ratio (1.20; 0.86–1.21). Triglyceride (85; 40–149 mg/dl) and fibrinogen (294; 170–410 mg/dl) levels were within normal limits. Cytomegalovirus antigenaemia was not detected. Epstein Barr virus (EBV) viral capsid antigen antibody (VCA) was negative for IgM and positive for IgG. Computed tomography (CT) of the chest and abdomen demonstrated mediastinal lymphadenopathy as well as hepatomegaly. A skin biopsy from the right lower thigh exhibited lobular panniculitis with mixed inflammatory infiltrations of neutrophils, lymphocytes and histiocytes without any atypia (Fig. 2a, b). Some histiocytes had phagocytized erythrocytes (Fig. 2c). A bone marrow biopsy revealed hypercellularity with increased numbers of megakaryocytes and granulocytes (Fig. 2d). Chromosomal analyses of bone marrow cells showed 46, XY, t(7; 17) (q36; q21). Bone marrow blasts accounted for 1% of the cells. Based on clinical, histopathological and molecular analyses, the patient was diagnosed as having CHP with very low-grade MDS. He required erythrocyte transfusion and erythropoietin therapy for his low haemoglobin level. He was treated with a low-dose oral corticosteroid (betamethasone 1 mg daily) because he chose not to have aggressive therapy from among a variety of treatment options at the first diagnosis. However, bacterial pneumonia led to the occurrence of disseminated intravascular coagulation (DIC) and he died 3 months after the initial examination. No autopsy was performed.
Fig. 1. Erythematous plaques (a) on the arm and (b) on the leg.
Fig. 2. (a) A biopsy specimen from the leg (haematoxylin and eosin (HE), original magnification ×20). (b) Inflammatory cells in square of Fig. 2a (HE, ×200). (c) Phagocytosed erythrocytes (arrows) (HE, ×400). (d) A bone marrow biopsy finding (HE, ×200).
CHP was originally described by Winkelmann & Bowie in 1980 (1). The main features of CHP include fever, multiple subcutaneous nodules, hepatosplenomegaly, liver function abnormalities, cytopaenia and intravascular coagulation. Histopathologically, skin lesions show infiltration of benign histiocytosis with phagocytosis of erythrocytes, leukocytes and platelets in subcutaneous tissue. The case reported here had these main features, and thus corresponded histopathologically to CHP. Although we did not examine the T-cell receptor gene clonality in the skin biopsy specimen, subcutaneous panniculitis-like T-cell lymphoma was excluded due to definite lack of atypical lymphocytes in the skin. According to a previous report, treatment of CHP includes glucocorticoids in combination with cyclosporine and combined chemo-therapeutic medications (2). However, recalcitrant cases of haemorrhagic complications, such as DIC, have been reported (3, 4). Although CHP has an uncertain aetiology, there is a pathophysiological similarity between CHP and haemophagocytic lymphohistiocytosis. The latter is a reactive process to a multitude of disorders, such as infectious and neoplastic conditions, featuring haemophagocytosis by proliferating benign histiocytes in the lymphoreticular system. The diagnostic criteria of haemophagocytic lymphohistiocytosis requires at least 5 of 8 criteria, including fever; splenomegaly; cytopaenia; hypertriglyceridaemia and/or hypofibrinogenaemia; haemophagocytosis in the bone marrow, spleen, or lymph nodes without evidence of malignancy; low or absent NK-cell cytotoxicity; hyperferritinaemia; elevated soluble IL-2Rα chain (≥ 2,400 U/ml). However, the current case did not satisfy the diagnostic criteria of haemophagocytic lymphohistiocytosis, namely the patient had only 2 of 8 criteria including fever and hyperferritinaemia (5). In contrast to haemophagocytic lymphohistiocytosis, CHP shows a benign histiocytic infiltration with haemophagocytosis in the subcutaneous tissue, resulting in the appearance of subcutaneous nodules. Thus, CHP is sometimes treated as regional skin lesions of haemophagocytic lymphohistiocytosis and embedded in a continuum (6).
While there were some cases in which CHP progressed to T-cell or B-cell lymphoma (7, 8), there has been no report of CHP associated with MDS. When a dermatological clinician is presented with a histopathological finding of haemophagocytosis in subcutaneous tissue, it is important to suspect CHP and perform bone marrow examination.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize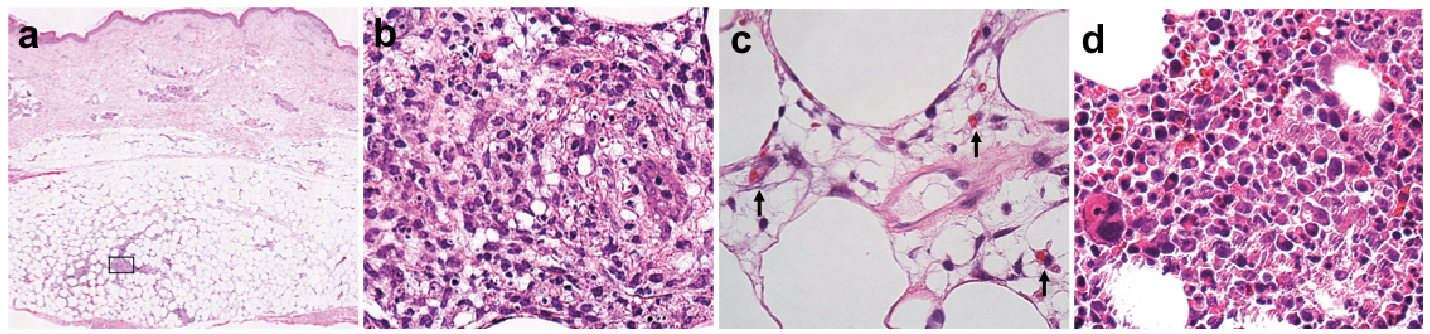 Click to show fullsize
Click to show fullsize