


1School of Medicine, Pontificia Universidad Católica de Chile, 2National Institute of Rehabilitation, Pedro Aguirre Cerda, 4Departamento de Neurología y Neurocirugía, Hospital Clínico Universidad de Chile y Programa de Anatomía y Biología del Desarrollo, Facultad de Medicina, Universidad de Chile, 3Department of Pediatrics, Neurology Unit, Clínica Las Condes, Lo Fontecilla 441, Santiago, Chile, 5Neuromuscular and Neurogenetic Disorders of Childhood Section, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, USA, and 6Unit of Neuromuscular and Neurodegenerative Disorders, Department of Neurosciences, IRCCS Bambino Gesu? Children’s Hospital, Rome, Italy. E-mail: ccastiglioni@clc.cl
Accepted Aug 22, 2016; Epub ahead of print Aug 26, 2016
Collagen VI-related muscular dystrophies (COL6-RD) comprise a continuous spectrum of clinical severity, ranging from the most severe phenotype Ullrich congenital muscular dystrophy (UCMD), through intermediate phenotypes, to a milder Bethlem myopathy (BM) (1). These inherited diseases are caused by autosomal dominant or recessive mutations in the genes COL6A1, COL6A2 and COL6A3, which encode the main 3 α-chains of collagen VI, a component of the extracellular matrix (ECM) that is present in the vast majority of connective tissues and is implicated in its organization (2). The prevalence of COL6-RD is estimated as 0.77:100,000 in BM and 0.13:100,000 in UCMD (3). Clinical features are secondary to the dysfunction of the collagen VI in the ECM of muscle and connective tissues of tendons, subcutaneous and dermal layers of the skin (1, 2). BM is characterized by slow progressive weakness of the proximal muscles and contractures that characteristically involve multiple joints. Although the course of the disease is slowly progressive, many patients exhibit a marked decrease in muscle strength between the 4th and 5th decades, with approximately half of them becoming wheelchair-dependant after the 5th decade (1, 2). Cutaneous manifestations include: presence of keratosis pilaris, mostly in the extensor surfaces of proximal legs and arms; formation of abnormal scars, especially keloids, either spontaneous or after minor trauma; rough or dry skin; striae rubrae; cigarette paper scars; and, in younger patients with UCMD, soft velvety skin on the palms and soles (4–7). Preclinical studies have observed decreased tensile strength of the skin and altered collagen fibril and basement membrane architecture in Col6a1 null mice (8).
We report here a case of spontaneous formation of keloids in a previously non-diagnosed BM patient who had 3 daughters also displaying features of BM.
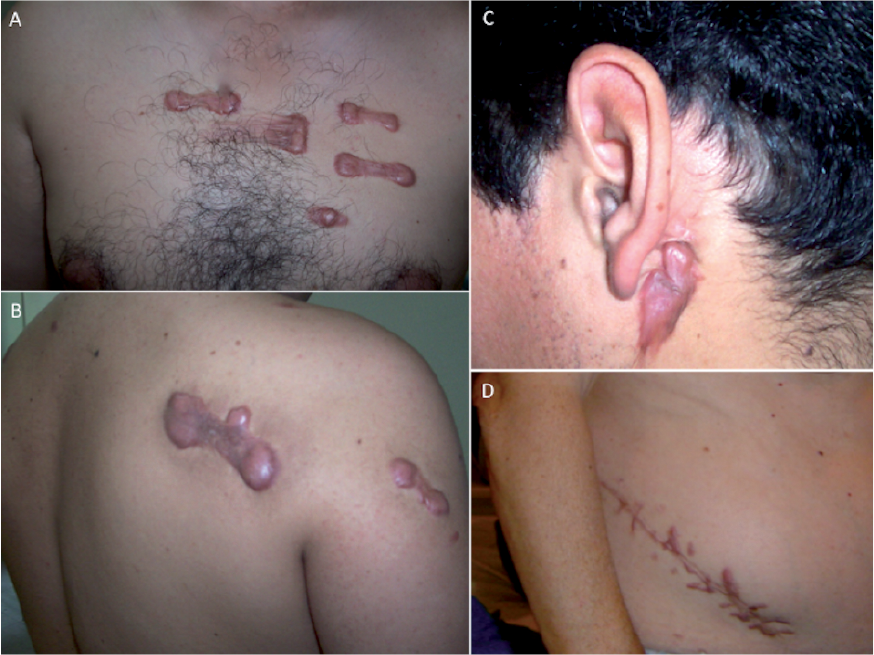
The patient, a male aged 45 years, was born with irreducible congenital torticollis, needing surgery to release the contracture. He had normal development with no delay in motor milestones and did not report any difficulties until the third decade of life. After the age of 25 years, he perceived mild difficulty in climbing stairs and standing up from the floor. Physical examination revealed mild proximal limb and facial weakness, positive Gowers’ sign, indicating signs of proximal muscle weakness, and contractures of the long finger flexors. Skin examination revealed prominent keloids due to mild traumas and itching lesions in the sternal area, upper back and the lateral side of the right arm. He also has a large keloid scar secondary to the torticollis release surgery (Fig. 1).
Fig. 1. Keloid formation in Bethlem myopathy. (A) Prominent keloids subsequent to itching lesions on the sternal area. (B) Spontaneous keloids in the upper back and lateral face of the right arm. (C) Retro-auricular keloid following torticollis release surgery. (D) Keloid following nephrectomy in the patient’s grandfather.
The patient’s youngest daughter, age 14 years, was born with hypotonia and multiple congenital contractures at the knees, hips and elbows, and congenital hip dislocation. Most contractures remitted at 6 months with physiotherapy. She achieved independent walking at 15 months. Physical examination revealed mild proximal weakness, mild scoliosis and no contractures. In activities of daily living, she reported difficulty in climbing stairs and pain in the lower limbs after physical activity. Muscle phosphocreatine kinase was slightly elevated, 260 U/l (normal range 38–176 U/l), and a muscle biopsy showed only mild myopathic changes. The patient’s middle daughter, aged 18 years, was born with congenital torticollis, and presented delayed motor milestones. She had difficulty running and was unable to complete sport activities at school. Physical examination revealed moderate proximal weakness with positive Gowers’ sign and proximal joint contractures. The patient’s eldest daughter, aged 24 years, was born with a congenital hip dislocation and developed scoliosis and difficulty playing sports at school. The 2 daughters have distal hyperlaxity, and keratosis pilaris on the arms; neither have keloid formation (Fig. S1).
Their paternal grandfather, who is over 60 years of age, had a large keloid in the incision site from a nephrectomy (Fig. 1). He has never sought medical advice, despite a clinical history of proximal weakness that began around his 5th decade, with difficulty climbing stairs associated with an increased risk of falls, which limits him in going out of the house. None of the patients had ever had prominent acne and their skin was not darkly pigmented.
Considering the clinical picture of the family suggestive of a dominantly inherited BM, patients were genotyped for mutations in collagen VI. Genetic study showed a missense mutation in COL6A2 (c.820 G>A, p.Gly268Ser) that causes a glycine substitution in the Gly-X-Y collagenous motif, at the beginning of the collagenous triple helical domain. The c.820 G>A mutation segregated in all the affected patients. This mutation has not been reported previously, but is predicted to be a typical dominantly acting missense mutation in collagen VI (9).
Keloids are a multifactorial disorder characterized by an abnormal response to tissue regeneration. Genetic susceptibility has been postulated by its prevalence in certain ethnic groups, family history and similarity between twins (10). The appearance of spontaneous keloids has been reported in very few genetic diseases, and there is a small group of congenital disorders with a high predisposition to spontaneous formation of keloids, such as Rubinstein-Taby, Dubowitz, Noonan, Goeminne syndromes and conjunctivo-corneal dystrophy (11, 12). It should be noted that this may also be a characteristic feature of COL6-RD (7). The relationship between collagen VI and keloids can provide important additional information about the pathogenesis of keloid formation. It has been shown that the ultrastructure of dermal collagens in patients with UCMD is altered as a consequence of collagen VI mutations (13). Type I and VI collagen gene expression is activated in keloids showing excessive levels of collagen VI and I and/or an increase in the collagen I/collagen III ratio in keloids, which it is suggested is triggered by elevated expression of transforming growth factor-β (14). Collagen VI is a critical component in maintaining the integrity of the skin, promoting roles such as organization and communication between cells. Recently, it has been found that collagen VI regulates the organization and composition of de novo ECM within the skin, suggesting a key role in wound healing and tissue repair (15). Therefore, its alteration appears to make skin susceptible to a variety of abnormalities, including spontaneous keloids, which are part of the clinical spectrum of skin alterations related to COL6-RD.
Until now, the pathogenic connection between collagen VI disorders and spontaneous formation of keloids has not been clearly established, nor is it known whether the degree of propensity for keloid formation correlates with other aspects of the disease, such as contracture formation.


 Click to show fullsize
Click to show fullsize