


1Department of Dermatology, Toulouse University Hospital, Toulouse, 24 chemin de Pouvourville TSA 30030, FR-31059 Toulouse cedex 9, 2U 1056 INSERM, FRE 3742 CNRS, Université Toulouse III ‘Différenciation Epithéliale et Autoimmunité Rhumatoïde’Place du Dr Baylac, 3Anatomo Pathology Department, IUC Oncopole, and 4Medical Genetics Department, CHU Purpan, Paul Sabatier University, Toulouse, France. E-mail: maella.severino@hotmail.fr
Epidermolytic ichthyosis (EI) is a rare disorder of keratinization belonging to the group of keratinopathic ichthyosis. EI is an autosomal dominant disease due to mutations in the genes encoding keratin 1 (KRT1) or keratin 10 (KRT10) expressed in the suprabasal layers of the epidermis (1). EI is characterized by erythroderma, blistering and erosions at birth, followed by generalized hyperkeratotic and verrucous lesions from early childhood. The lesions are generalized and palmo-plantar keratoderma may be seen (2). EI is a severe disease due to skin aspect, itching and recurrent episodes of skin infections with malodorous skin. Histological examination of skin lesions shows thickening of the stratum corneum, vacuolar degeneration of the epidermal suprabasal layer, and some clumping of tonofilaments in the keratin. Whereas EI is in the form of generalized skin lesions, localized segmental lesions along the Blaschko’s lines have been reported rarely as a consequence of post-zygotic somatic mutations in KRT1 or KRT10. Post-zygotic mosaicism in EI must be distinguished from epidermolytic naevi, a naevoid variant of keratinopathic ichthyosis characterized by localized hyperkeratotic lesions present at birth without an initial phase of blistering (3, 4). Post-zygotic mosaicism in EI usually has a limited distribution. We report here 2 cases of EI with an extensive distribution.
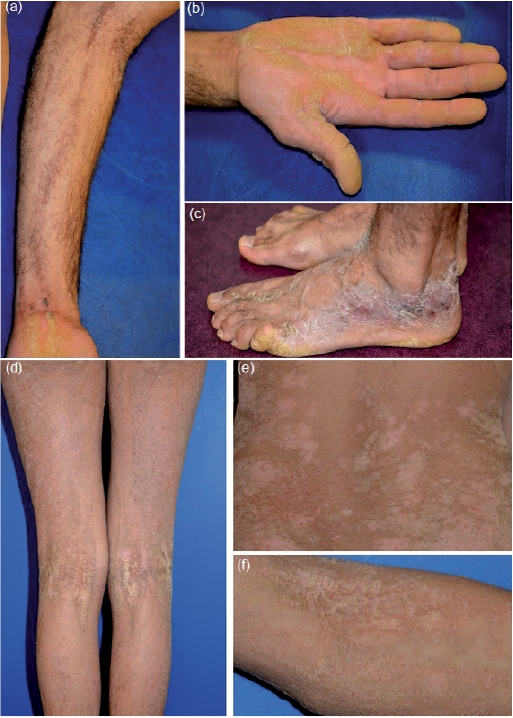
Two patients (1 male, 1 female, aged 30 and 17 years, respectively), with unaffected parents, presented at birth with blisters, erosions and erythroderma. They developed extensive, but not generalized, hyperkeratotic lesions distributed along the lines of Blaschko, involving 40% and 80% of body surface area, respectively. The male patient had skin lesions located on the folds, as well as on the trunk, limbs, palms and soles (Fig. 1a–c). The female patient had extensive verrucous plaques, which were more pronounced in the folds and back (Fig. 1d–f). She had no palmo-plantar involvement. Histopathological examination of her lesional skin showed major epidermolytic hyperkeratosis, sometimes with parakeratotic and degenerative lesions in the granular layer.
Fig. 1. Male patient: (a) Hyperkeratotic lesions distributed along the lines of Blaschko in the elbow fold and forearm. (b, c) Severe hyperkeratosis of the palms and foot. Female patient: (d, f) Hyperkeratotic lesions distributed along the lines of Blaschko in the elbow and knee folds. (e) More pronounced hyperkeratotic verrucous plaques located in the back and folds.
Sequencing of KRT1 and KRT10 genes was performed by the Sanger method. For the male patient, molecular analysis of lesional skin revealed the heterozygous mutation c.526_531delGTGAAG in exon 1 of KRT1. This mutation predicted a deletion of 2 amino acids in the encoded protein (p.Val176_Lys177del). It was also detected in leukocytes from peripheral blood, but the sequence corresponding to the mutant allele was in a lower proportion compared with the normal sequence (Fig. S1a). For the female patient, the mutation c.466C>T (p.Arg156Cys) in exon 1 of KRT10 was identified from peripheral blood leukocytes as well as from saliva. In both tissues, electropherograms showed that the mutated allele was in a lower proportion compared with the wild-type allele (we estimated that two-thirds of the cells carried this mutation). This was subsequently confirmed by next-generation sequencing using a targeted panel involved in ichthyosis (Fig. S1b). Among the 226 reads of the region, 84 (37.2 %) were found to bear the mutation.
Two types of segmental mosaicism of autosomal dominant skin disorders are described. In type 1 mosaicism (as in our patients), one allele of the involved gene is mutated during embryonic development. Type 2 originates from loss of heterozygosity occurring in a heterozygous embryo at an early developmental stage (5).
Molecular proof of mosaicism in EI has been reported previously in 2 individuals (6, 7). The neonatal inflammatory phase was probably present in 2 cases, but was mentioned for one only of them (7). One had limited lesions, whereas the other had a very extensive distribution similar to our female patient (6). One of these 2 patients had molecular analysis from skin and lymphocytes that revealed KRT10 mutation (6).
The KRT10 mutation found in our female patient was previously described in a few patients with classic EI, but not in cases of mosaicism, whereas the KRT1 mutation found in our male patient was novel. This latter mutation is located at the end of the head domain of KRT1, just upstream of the highly conserved helix initiation peptide, a functional domain necessary for assembly of keratin intermediate filaments. Moreover, missense mutations at position 177 (p.Lys177Asn) or in a neighbouring amino acid (p.Ser178Pro, p.Arg179Pro) have been described as causative mutations (8–10). In addition, the p.Val176_Lys177del mutation was absent from the ExAC database. Thus, the p.Val176_Lys177del mutation is probably responsible for the phenotype.
Mutations reported in classic EI were described in 3 series (2, 11, 12) (25 different mutations from 56 patients). The majority of mutations concerned KRT10, the most frequent being the mutation p.Arg156His (c.467G>A). Except for palmo-plantar involvement, which is mainly associated with KRT1 mutations (3), no genotype-phenotype correlation was identified in EI.
The percentage of KRT1 or KRT10 de novo mutations is very high, from 50% to 75% (2, 11). Part of these de novo mutations may correspond to parental mosaic mutations, which are often undetectable using the classical Sanger method, since the mutant allele is present at a low intensity (7). Next-generation deep-sequencing methods offer opportunities to assess mosaicism, including the low-grade and should therefore permit the diagnosis of more cases of mosaicism in the future (13).
The 2 clinical cases described here were particular because of their extensive distribution, which made the diagnosis very difficult since they closely mimic a classical form of EI. The extent of skin lesions does not appear to be correlated with the mutation rate in leukocytes. Some authors have suggested that extension is linked to an early occurrence of the mutation that can affect both mesodermal germline and ectodermal tissues (14).
Recognition of a mosaic form of EI is important for genetic counselling, since there is a risk of transmission of a generalized form to the offspring, due to the possible presence of the mutation in the germ-line. A distinction between type 1 and 2 mosaicism is also important. In type 1 mosaicism, the risk of transmitting the gene to the next generation is dependent on the proportion of mutated germinal cells, whereas in type 2 this risk is 50% (5). In our cases, the exact risk cannot be determined precisely, but should be lower than the predicted transmission of 50% characteristic for autosomal dominant diseases. In conclusion, it is important to be aware of KRT1 or KRT10 mosaicism in an extensive distribution, in order to perform molecular analysis using an appropriate method and to provide appropriate genetic counselling.


 Click to show fullsize
Click to show fullsize