


1Department of Dermatology, Saint-Eloi Hospital, University Hospital of Montpellier, 80 avenue A. Fliche, FR-34295 Montpellier cedex 5, 2INSERM U1058, Montpellier, and 3Department of Medical Genetics, Purpan Hospital, University Hospital of Toulouse, Toulouse, France.
E mail: d-bessis@chu-montpellier.fr
Accepted Oct 26, 2016; Epub ahead of print Oct 27, 2016
Fibroblast growth factor receptor 3 epidermal naevus syndrome (FGFR3-ENS), also known as Garcia-Hafner-Happle syndrome, is a rare and distinctive epidermal naevus (EN) syndrome, clinically characterized by a systematized keratinocytic EN of soft and velvety type, with inconstant cerebral and skeletal involvement (1). We report here a new case of FGFR3-ENS with the postzygotic FGFR3 p.Ser249Cys mutation detected in both affected skin and urothelial cells.
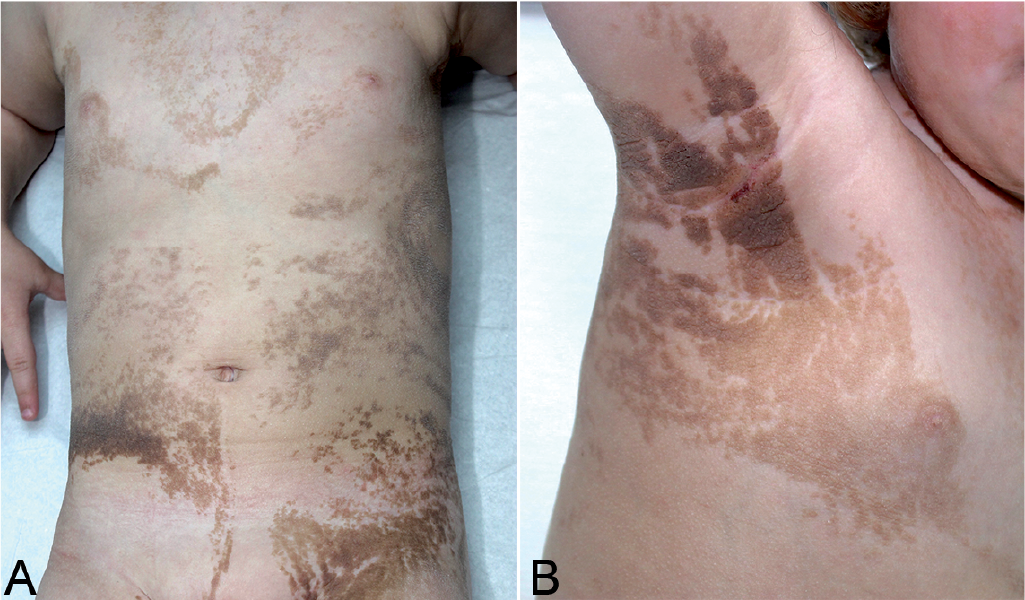
A 30-month-old girl was referred for assessment of systematized EN. She was born at term and was otherwise healthy, with normal skeletal and neurocognitive development and no remarkable family history. Skin changes were first noted on the neck at 4 months, with spreading to trunk, limbs and face over the first year of life. Physical examination disclosed a systematized and widespread linear brown EN of a soft and velvety type, following Blaschko’s lines, associated with confluent thick and papillomatous plaques over the neck, axillary and groin regions (Fig. 1). Histological examination of the EN revealed hyperkeratosis, acanthosis and papillomatosis. Urine sediment failed to reveal haematuria. Ophthalmologic examination, electroencephalogram, total-body skeletal radiographs, and pelvic ultrasonography were normal. Cerebral magnetic resonance imaging (MRI) showed bilateral cortical dysgyria of the anterior and basal temporal areas, with thinning of the median part of the corpus callosum.
Fig. 1. Systematized and widespread epidermal naevus distributed along Blaschko’s lines. (a) Trunk involvement. (b) Confluent papillomatosis acanthosis nigricans-like lesions of the axillary area.
After obtaining informed parental consent, DNA was directly extracted from a biopsy specimen of the skin lesion and adjacent normal skin, a blood sample, urine sediment, and scalp hair roots and saliva. The FGFR3 mutation c.746C>G (p.Ser249Cys) was identified in mosaic form in EN tissue, urothelial cells, scalp hair roots and saliva, but not in blood leukocytes or normal skin. Additional analyses using Minor Variant Finder software and allele-specific PCR amplification helped to refine the mosaicism in EN tissue, urothelial cells, scalp hair roots and saliva at 30%, 10%, 5% and 5%, respectively (Fig. 2).
Fig. 2. Detection of the c.746C>G (p.Ser249Cys) FGFR3 mutation in the proband by (A) allele-specific PCR and (B) Sanger sequencing. (A) Genomic DNA was isolated from different types of tissue and subjected to duplex PCR amplification generating 2 amplicons: a first amplicon of 397 bp used as an internal control (to check the efficiency of the amplification), and a second smaller amplicon whose reverse primer is located on the c.746C>G mutation (expected size 237 bp). This allele-specific primer allows efficient discrimination of the mutation in cases of mosaicism. The PCR products were then separated by electrophoresis in 2% agarose gel. Note the single 397 bp amplicon in the healthy control (gDNA isolated from blood) in Lane 1 vs. 2 amplicons with similar intensity in the heterozygous control for the c.746C>G mutation (trophoblast cells) in Lane 2. Lane 3: presence of the 2 expected amplicons with lower intensity for the smaller amplicon in the proband skin lesion specimen. In this tissue, the c.746C>G mosaicism was estimated at approximately 30%. Lane 4: absence of the mutation in the proband healthy skin specimen. Lane 5: presence of the 2 expected amplicons in the proband urine sample with mosaicism estimated at 10%. Lane 6: absence of the mutation in the proband blood sample. Lane 7: presence of the 2 expected amplicons in the proband saliva sample: mosaicism was estimated at approximately 5%. Lane 8: although very faint, the 2 expected amplicons can be noted in the proband hair sample with mosaicism estimated at approximately 5%. Lane 9: negative control. Lane 10: pBR322 DNA/BsuRI (HaeIII) DNA ladder (Thermo Fisher Scientific, Waltham, MA, USA). (B) Electropherograms generated by Minor Variant Finder (MVF) software to detect the c.746C>G mosaicism in the FGFR3 gene sequencing of, respectively: (a) the proband skin lesion specimen, (b) the proband urine sample, and (c) a negative control. After removing the background noise signal due to sequencing, the c.746C>G mosaicism was estimated by the MVF software at: (a) 34.2%, (b) 8.6%, and (c) 0% (no detection of the mutation) on the respective forward sequences. Note, this mosaicism rate was also calculated on the reverse FGFR3 sequence and the mean of the 2 ratios (forward and reverse) was calculated. The position of the c.746C>G mutation detected by the software is indicated by a vertical blue line.
These results were consistent with the diagnosis of FGFR3-ENS due to extensive somatic mosaicism linked to the p.Ser249Cys mutation in the FGFR3 gene.
One previous case of FGFR3-ENS related to the p.Ser249Cys FGFR3 mutation has been reported, similar to our observation of systematized bilateral EN, but without obvious clinical neurological or skeletal involvement (2). Our result confirms that the origin of FGFR3-ENS is not restricted to the recurrent p.R248C mutation.
The occurrence of urothelial cancer (UC) of the urinary tract or bladder in patients with ENS has been reported in 6 cases (3–5). The UC-EN association does not appear to be fortuitous based on: (i) the uncommonly premature age (16–24 years) for the development of UC; (ii) the common low-grade papillary subtype of UC; and (iii) the high prevalence of the genitourinary tract as a site of all non-skin cancers associated with EN (5). The potential involvement of oncogenic genes, such as FGFR3, HRAS and PIK3CA, in the pathogenesis of both EN and UC raises the suspicion of a common genetic basis with the presence of post-zygotic mosaicism in skin and urothelium. To our knowledge, only one observation has confirmed this hypothesis, in a patient with a Costello syndrome mosaicism, where the p.Gly12Ser HRAS mutation was detected in the tissue of both widespread EN and UC, as well as in non-neoplastic urothelium and blood leukocytes. Mutations of FGFR3, especially p.Ser249Cys (6), play a major role in the development of low-grade papillary UC and in numerous cases of EN. In our patient, the identification of the embryonic oncogenic FGFR3 mosaicism in the urothelial cells may account for the increased risk of UC in this type of EN. Moreover, it raises the question of the interest of a medical follow-up in patients with FGFR3-ENS, especially for urine sediment and urinary tract ultrasonography.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize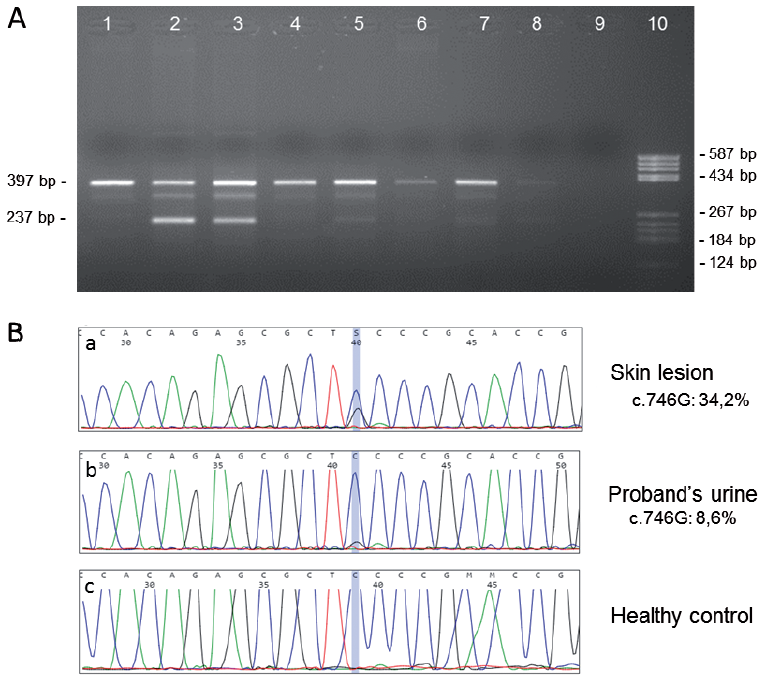 Click to show fullsize
Click to show fullsize