


1Department of Dermatology, 2Centre for Child and Adolescent Medicine, Divisions of General Pediatrics, Neuropediatrics, and Metabolic Medicine and 4Institute of Pathology, University Hospital, Im Neuenheimer Feld 440, DE-69120 Heidelberg, Germany, 3Department of Dermatology, Division of Immunology, Allergy and Infectious Diseases (DIAID), Medical University of Vienna, Vienna, Austria. E-mail: mona.bidier@med.uni-heidelberg.de
#Both authors contributed equally.
Accepted May 17, 2018; Epub ahead of print May 18, 2018
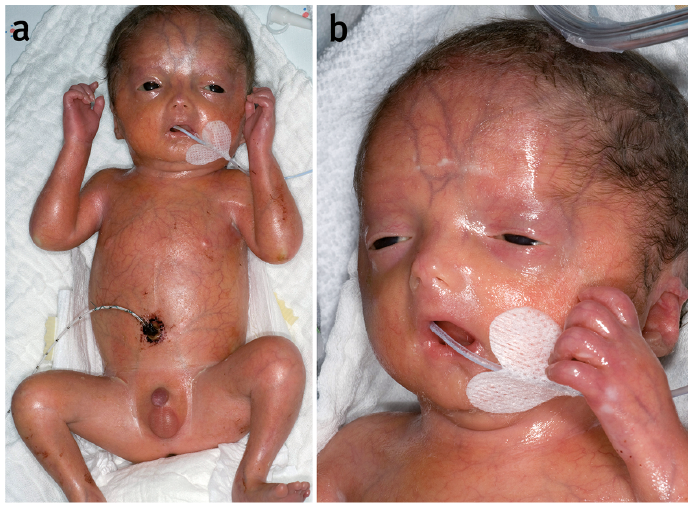
We report here 4 patients with restrictive dermopathy (RD) who presented in the paediatric and dermatology department of the University of Heidelberg from 1996 until 2017. All of the patients were premature infants displaying the pathognomonic clinical picture of RD: tense, vulnerable and translucent skin, superficial erosions, joint contractures, reduced motoricity and a typical facies with a small pinched nose, mouth fixed in an o-position, low-set ears and micrognathia (Fig. 1). Skin biopsies for light and electron microscopy showed collagen bundles parallel to the skin surface and a hypoplasia of appendages as common pathological findings. In 2 biopsies, the collagen fibres were small and in 3 of the 4 patients the rete ridges were flattened and elastic fibres were reduced or arranged in tiny clumps (Fig. 2b). All 4 babies developed respiratory insufficiency and died between days 6 and 32 after birth. (For more detailed information on each case see Table SI).
Fig. 1. Clinical presentation. The patient’s skin appeared tense, vulnerable and translucent with clearly visible vessels, scaling and erosions (case 1). In addition, the joints were contractured and the patient showed a reduced motoricity. There were typical facial dysmorphies: hypertelorismus, a small nose, low-set ears, micrognathia, and a small round open-mouth fixed in the o-position Permission is given by the parents to publish these photos.
Fig. 2. Structural skin changes in restrictive dermopathy (RD). Healthy newborn skin (a, c, e, g) and skin samples from a patient with RD (b, d, f, h) were studied. By light microscopy (10×), the epithelium in the skin sample from the patient with RD was slightly acanthotic and showed mild orthohyperkeratosis (b). The rete ridges were flattened, and the appendages were rudimental, whereby the hair shafts in some sections were without pathological findings, in others they were slightly rarified. The thickness of the corium was reduced. Electron microscopy of skin sections of a patient with RD (10,000×) (h) in contrast to healthy skin (9,000×) (g) showed many closely lying collagen bundles and few underdeveloped elastic fibres. Immunohistochemical staining identified elastin expression in skin tissue of (c) a healthy newborn 1 day old (20×). In skin tissue of a newborn with RD (patient 4), age 2 days (gestational age: 29+5 weeks), elastin expression presented with a diffuse dermal distribution of the fibres (20×) (d) (anti-elastin antibody, Abcam (ab21610)). Fibulin expression in skin of a healthy newborn 1 day old (e) was readily detectable, while there was little evidence for fibulin expression (20×) (f) in skin tissue of a newborn with RD (patient 4), 2 days old (gestational age: 29+5 weeks) (anti-fibulin 1 antibody, Abcam (ab54652)).
RD has a pathognomonic phenotype; however, the underlying structural skin changes remain to be defined. One possible explanatory approach may be a developmental arrest of extracellular matrix components, e.g. elastic fibres. As this hypothesis has not yet been addressed, immunohistochemical analysis of elastic fibres was performed in healthy newborns and those with RD.
RD is a rare, lethal, autosomal recessive genodermatosis, first described as a distinct entity in 1986 by Witt et al. (1), whereas the first clinical reports by Antoine (2) allegedly date back to 1929. Navarrho et al. identified the disease as a laminopathy in 2004 (3). Laminopathies include different diseases, such as Hutchinson-Gilford progeria syndrome, mandibuloacral dysplasia, and dilatative cardiomyopathy. They are based on defects of the nuclear membrane protein lamin A. The main cause of RD is an autosomal recessive gene defect of the ZMPSTE24 gene. There are different mutations observed in patients with RD, but the most common one is c.1085_1086 in exon 9 (4). The mutations identified to date lead to a loss of function of the zinc-metallo-proteinase ZMPSTE24. As a result, the nuclear-membrane precursor protein prelamin A cannot be completely processed to lamin A (5, 6), leading to a loss of mature lamin A and an increase in the intermediate stage farnesylated prelamin A. This intermediate stage of lamin A accumulates at the nuclear rim, causes misshapen nuclei (7–9) and has an intrinsic toxic effect on cells (7, 10–12).
RD has a characteristic clinical picture, as seen in our 4 patients. Additional findings are skeletal abnormalities, including clavicular hypoplasia and abnormalities in the long bones. The mean gestational age is 31 weeks, and foetal dyskinesia is present in all reported cases (13). Affected patients have lung hypoplasia and inspiratory dysfunction due to thoracic stiffness. Respiratory insufficiency is the most common cause of death in patients with RD. Some patients are stillborn, and most patients die within the first days of birth. The longest reported survival is 120 days (14). Currently, there is no curative therapy for RD. Therefore, prenatal diagnosis by chorionic villus biopsy or amniocentesis and providing genetic advice to affected families play an important role. Histological findings of the skin are a thick epidermis with hyperkeratosis, coarse keratohyaline granules, absent rete ridges, a relatively thin dermis with collagen fibres oriented parallel to the epidermis, decreased elastic fibres, hypoplastic appendages, and a relatively thick subcutaneous adipose tissue. Electron microscopy shows abnormalities of keratin filaments, keratohyaline granules, and more pronounced collagen and elastic fibres (13).
We looked for evidence of a misguided development of elastic fibres in RD. Electron microscopy showed closely lying collagen bundles and few, underdeveloped elastic fibres in RD (Fig. 2 h) compared with normal skin (Fig. 2 g). Staining for elastin revealed elastin stretched fibres in healthy skin, while in RD the fibres appeared coiled and twisted (Fig. 2 c, d). Fibulin expression was found in healthy skin, but was hardly detectable in a newborn with RD (Fig. 2 e, f). Expression analysis suggests that there is an aberration in the development of elastic fibres, especially concerning elastin, leading to a diffuse distribution of these fibres in the skin of patients with RD. This disordered distribution pattern may be one explanation for the tightened skin in patients with RD.
The authors thank Professor Manfred Wehnert (Institute of Human Genetics, University Hospital Greifswald) for performing the genetic analysis.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize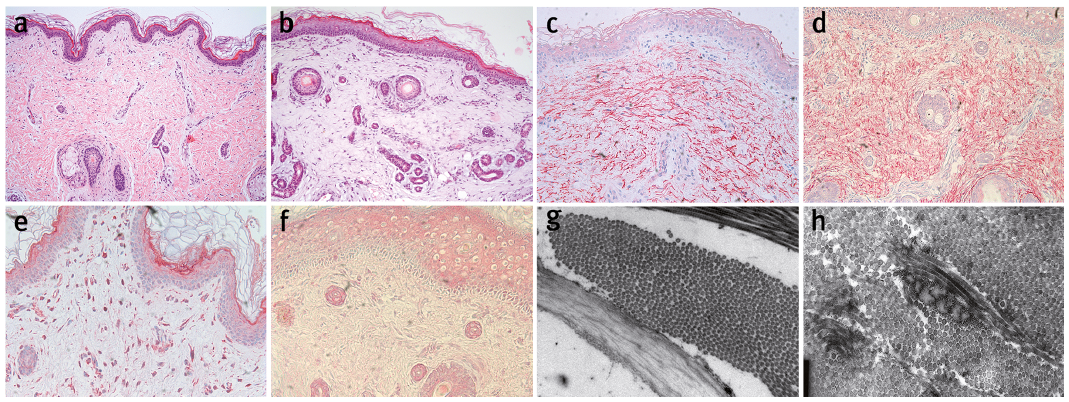 Click to show fullsize
Click to show fullsize