


1Galician Public Foundation of Genomic Medicine (FPGMX)-SERGAS, Genomics Medicine Group-USC, CIBERER, IDIS, 2Department of Forensic Sciences, Pathological Anatomy, Gynecology, Obstetrics and Pediatrics, USC, 3Dermatology Service of University Clinical Hospital of Santiago, Santiago de Compostela, 4Dermatology Service of University Clinical Hospital of Vigo, Vigo, 5Dermatology Service of Albacete University Hospital Complex, Albacete, 6Dermatology Service of University and Polytechnic Hospital La Fe, Valencia, and 7Clinical Genetics Service of Central University Hospital of Asturias, Oviedo, Spain
Autosomal recessive congenital ichthyosis is a group of rare non-syndrome diseases that affect cornification. PNPLA1 is one of the 12 related genes identified so far. Mutation screening of this gene has resulted in the identification of 13 individuals, from 10 families, who carried 7 different PNPLA1 mutations. These mutations included 2 missense, 2 frame-shift and 3 nonsense, 3 of them being novel. One of the identified variants, c.417_418delinsTC, was highly prevalent, as it was found in 6 out of 10 (60%) of our autosomal recessive congenital ichthyosis families with PNPLA1 mutations. Clinical manifestations varied significantly among patients, but altered sweating; erythema, palmar hyperlinearity and small whitish scales in flexor-extensor and facial areas were common symptoms. Haplotype analyses of c.417_418delinsTC carriers confirmed the existence of a common ancestor. This study expands the spectrum of the PNPLA1 disease, which causes variants and demonstrates that the c.417_418delinsTC mutation has founder effects in the Spanish population.
Key words: ARCI; Spanish population; PNPLA1; founder effects; c.417_418delinsTC.
Accepted May 22, 2019; E-published May 23, 2019
Acta Derm Venereol
Corr: PhD Ana Vega, Molecular Medicine Service, Fundación Pública Galega de Medicina Xenómica, University Hospital Santiago de Compostela, floor -2, Santiago de Compostela, Spain. E-mail: ana.vega@usc.es
Due to the growing importance of PNPLA1 mutations in the development of autosomal recessive congenital ichthyosis (ARCI), we decided to sequence this gene in a large, well-characterized cohort of Spanish ARCI patients. The mutational analysis revealed 7 different PNPLA1 mutations, 3 of which were novel, in 13 individuals from 10 families. Interestingly, one of the identified mutations was present in 60% of the families (c.417_418delinsTC; p.Ser140Pro). Haplotype analysis of the c.417_418delinsTC carriers de-monstrated that these families are descendants of a recent founder who lived in the XI century, implying that this recurrent mutation has founder effects in the Spanish population.
Numerous hereditary cornification disorders that are clinically and aetiologically heterogeneous and follow a Mendelian inheritance pattern are grouped under the term of inherited ichthyosis. Autosomal recessive congenital ichthyosis (ARCI) are a subgroup of non-syndrome ichthyosis, whose phenotypic spectrum ranges from harlequin ichthyosis (HI; OMIM 242500), to less severe phenotypes such as congenital ichthyosi-form erythroderma (CIE; OMIM 242100) or lamellar ichthyosis (LI; OMIM 242300) the last two being the most common phenotypes. To date, 12 genes have been implicated in the development of ARCI: ABCA12, ALOX12B, ALOXE3, CYP4F22, NIPAL4, TGM1, LIPN and more recently CERS3, PNPLA1, CASP14, SDR9C7 and SULT2B1 (1–3).
PNPLA1 is a gene that encodes a 58 kDa protein involved in the glycerophospholipid metabolism of the cutaneous barrier. Forty-seven disease causing mutations have been described in PNPLA1 so far (4–12), the majority of which were reported within the last two years (4–9, 11, 12). Due to the growing importance of PNPLA1 mutations in the development of ARCI, we decided to screen for PNPLA1 defects in a large, well-characterized cohort of individuals with ARCI. Screening has resulted in the identification of a recurrent variant, c.417_418delinsTC, which was present in approximately half of our families. Given the high prevalence of this mutation among our Spanish patients, we tested the hypothesis that this mutation arose in a founder ancestor in the distant past.
Thus, the aims of this study are: (i) to report new causative PNPLA1 mutations in a Spanish cohort of patients with ARCI, (ii) to computationally assess the effect of the recurrent c.417_418delinsTC mutation on protein function, (iii) to evaluate whether families who carry this variant share a common ancestor and, if so, (iv) to estimate the time to their most recent common ancestor (TMRCA) and the age of the mutation.
A cohort of 91 Spanish patients (81 different families) with clinical suspicion of ARCI was studied. A dermatologist clinically characterized all affected individuals. Clinical and genealogical information was collected for each family. Pedigrees and available clinical photos of the families are presented in Fig. 1 and Fig. S1. This study, carried out at FPGMX, was approved by the Galician Ethical Committee for Clinical Research (Code 2013/056) and the procedures followed were in accordance with the Declaration of Helsinki. All participants provided written informed consent.
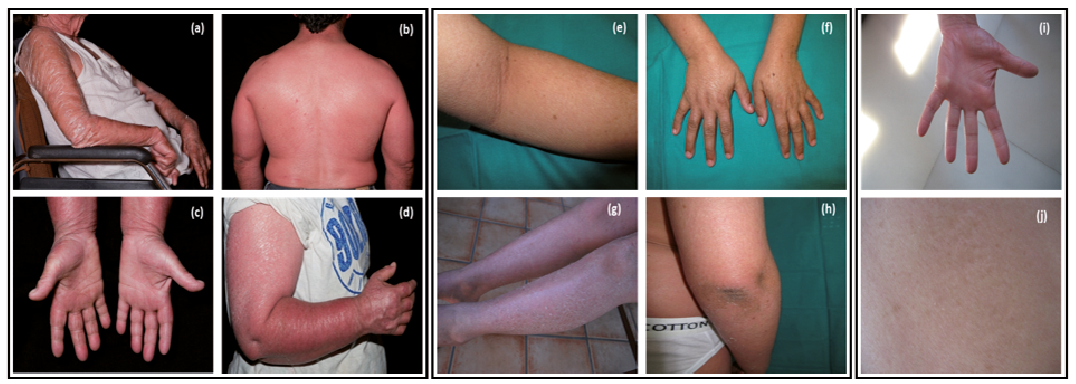
Fig. 1. Clinical photos of patients from families 23 (a–d), 62 (e–h) and 115 (i, j). a) Arms of the proband with generalized erythema and large adherent scales, b and c) back and hands of proband´s son with generalized erythema accompanied by fine white scales and palmar hyper linearity, respectively, d) proband´s daughter showing underlying erythroderma and generalized scaling, e) large whitish scales in legs with underlying erythema, f) dry scaly skin on hands, g) fine white scaling in extensor areas, h) very mild affected flexor surfaces, i) palmar hyper linearity, j) mild generalized scaling.
The PNPLA1 gene was examined by Sanger sequencing or targeted resequencing on SOLiD 5500xl or Ion Proton Platforms (Thermo Fisher Scientific; San Jose, CA, USA). Identified variants were confirmed in the proband and close relatives. The potential pathogenicity of each variant was assessed by using the Alamut® Visual 2.8.1 software (Interactive Biosoftware, Rouen, France).
The model published by Wilson et al. (13) was employed to represent the 3D human PNPLA patatin-like domain with the SWISS-MODEL tool (http://swissmodel.expasy.org). The effect of the mutation in the protein structure was evaluated with Swiss-PdbViewer (http://spdbv.vital-it.ch). The degree of conservation of the Ser140 was evaluated using the Clustal Omega tool (http://www.ebi.ac.uk/Tools/msa/clustalo).
Haplotypes of c.417_418delinsTC mutation carriers, and 80 Spanish healthy controls were reconstructed by genotyping 8 microsatellite markers. These markers spanned a segment of 10Mb flanking the PNPLA1 gene locus. The age of the mutation was calculated with DMLE+ (http://www.dmle.org). The TMRCA was estimated by applying different linkage disequilibrium algorithms. Further details are available in Appendix S1.
Ten families, 8 new and 2 previously reported (5, 14, 15), harboured 7 different PNPLA1 mutations. These families encompassed 8 patients with LI, 4 patients with CIE and one individual without any available phenotypic information. Patient´s clinical and genotypic data is presented in Table I. The 7 mutations included 2 missense, 2 frameshift and 3 nonsense mutations. Three of the 7 mutations identified were novel: c.282dup; p.(Lys95*), c.729C>G; p.(Tyr243*) and c.892C>T; p.(Arg298*). The 4 known mutations comprised of 2 frameshift [c.1143del; p.(Pro382Alafs*74) and c.820del;p.(Arg274Glyfs*8)] previously described by Zimmer et al. (4) and Pichery et al. (6), respectively, and 2 missense [c.100G>A; p.(Ala34Thr) and c.417_418delinsTC; p.(Ser140Pro)] previously reported in a Spanish (13) and French family (4). Characteristics of the mutations found in the present study are described in detail in Table SI. The most prevalent mutation, c.417_418delinsTC, was found in approximately half of the families (60%). Analysis of the protein structure showed that the Ser125 residue (structurally equivalent to Ser140 in PNPLA1) is present in a loop region, formed by the highly conserved residues (Fig. 2), indicating that the local environment of Ser140 has probable structural consequences.
Table I. Clinical manifestations, treatment, mutations and geographical origin of PNPLA1 mutation carrier patients
Fig. 2. Characteristics of the missense variant c.417_418delinsTC (p.Ser140Pro) in the PNPLA1 gene. (a) Comparison of partial amino acid sequence of human PNPLA1 across different species. Serine (S) within the red box indicates the conserved residue affected by the S140P mutation (b) nucleotide sequence in a homozygous affected member in the upper panel, a heterozygous carrier in the middle panel and wild type sequence in the lower panel, c) 3D-dimensional structure of the patatin domain indicating beta sheets in yellow, alpha helices in green, wild type Ser 125 and mutant Pro 125 in green colour, Arg151 in yellow and Phe 123 in orange. Green dots represent a strong H-bond while purple dots represent a clash (short distance repulsive energy).
Clinical manifestations varied significantly among patients, but altered sweating, erythema, palmar hyperlinearity and small whitish scales that affected flexor and extensor surfaces and facial areas were common symptoms. However, prematurity and dark scales were infrequent. Ectropion, collodion at birth, palmoplantar keratoderma, alopecia and big scales were occasionally seen. Seven out of 12 patients were taking topical retinoids while the rest were treated with a combination of oral and topical retinoids (Table I).
A clear correlation between the location/type of mutation and the resulting phenotype was not found. According to the data in Table I, the combination of nonsense and missense mutations led to LI pheno-types (Families 46, 98, 115), as well as frameshift and missense or homozygous frameshift variants (Families 62 and 137). The only carrier of a homozygous nonsense mutation showed CIE (Family 69). Patients with compound missense mutations had LI or CIE phenotypes indistinctly (Families 18, 23, 47, 119).
Reconstructed haplotypes of c.417_418delinsTC carriers are presented in Table SII. All carriers shared the same allele (7) for marker D6S439. A 4.1 Mb prevalent core haplo-type (5–7–5) ranging from markers D6S497 to D6S1548 was identified. This core haplotype was found on 4 out of 9 carrier chromosomes (44%) versus 18 out of 160 (11%) found on control chromosomes. To date the TMRCA of these 6 families, we used different algorithms, which resulted in different estimates. Averaging all these estimates would place the TMRCA approximately 38 generations ago (95% CI 29-46). However, the age of the c.417_418delinsTC, ranged from 103 to 108 generations, dating the first appearance of the mutation to approximately 2,575–2,700 years ago (assuming 25 years per generation). TMRCA and mutation age estimates are shown in Table SII.
According to the localization of the reported PNPLA1 mutations, Zimmer et. al (4) proposed a highly conserved, extended patatin domain that ranges from amino acid positions 1 to 288, increasing the size of the former functional region (Ile16-Thr185). Considering Zimmer´s proposal, 2 of the 3 novel mutations described in this study, c.282dup; p.(Lys95*) and c.729C>G; p.(Tyr243*) are located within this extended domain in exons 2 and 5, respectively. These variants create truncated mRNAs that would most likely be degraded by the process of nonsense-mediated decay. Our third novel nonsense mutation in exon 6, c.892C>T; p.(Arg298*), is the fifth variant reported, which is situated outside of the extended patatin domain (4, 5, 11). PNPLA1 protein contains a proline-rich domain (residues 326–451) in the C-terminal, which is conserved across species. Interestingly, all mutations located outside the extended patatin domain p.(Glu313Aspfs*49), p.(Pro370*), p.(Ser382Alafs*74), p.(Ala434Hisfs*10) and our novel variant p.(Arg298*) create premature stop codons before or within the proline-rich domain, with the exception of p.(Ser382Alafs*74). A schematic representation of PNPLA1, including novel and previously reported mutations, is shown in Fig. 3.
Fig. 3. Schematic representation of the PNPLA1 isoform 3 (NM_001145717) and the location of all mutations found to date including the novel reported in this study. The extended patatin-like domain of the gene is coloured in pink. The new mutations reported by this study are in bold letters while the founder Spanish mutation is underlined.
The 3D model of the PNPLA patatin-like domain showed that the c.417_418delinsTC mutation, which involves the substitution of 2 nucleotides (GT>CT), alters a conserved serine residue located in a beta sheet hairpin loop (Fig. 2), which has an impact on the close hydrogen-bond interactions with other residues affecting the protein structure.
The c.417_418delinsTC mutation has been described only in French and Spanish descents (4, 5). Due to the geographical proximity of these 2 countries, it is plausible that French and Spanish families share a common ancestor. The mutation could have arisen in Spain approximately 2,625 years ago (VII century BC) and brought to France by a Spanish carrier or vice versa.
To date, PNPLA1 mutations have been reported in different populations, French, Scandinavian, Iranian, Spanish, etc. (4, 10, 12, 14). The mutation frequency varies among the different populations, in Scandinavian countries mutations in PNPLA1 account for less than 1% of all ARCI-related mutations (10), whereas in highly consanguineous populations, such as Iran, the frequency reaches 15% (12). In our cohort, mutations in this gene represent 14% (13 out of 91) of the total ARCI cases. We propose that this unexpectedly high frequency of the mutated PNPLA1 could be due to the founder effect of the recurrent mutation, c.417_418delinsTC.
In conclusion, this study reports 3 novel mutations expanding the spectrum of the PNPLA1 disease causing variants and posits the possible mechanism underlying the pathogenicity of the c.417_418delinsTC mutation. Furthermore, we demonstrate that this mutation has founder effects in the Spanish population. TMRCA estimations suggest that carrier families could have descended from a single recent founder who lived in Spain during the XI century, while the mutation arose about 1,700 years prior.
The authors are grateful to families for their cooperation and to Jamie Allen for proofreading the manuscript and to Andrés Etxeita for the photos of the family 23. We also would like to thank the Spanish Association of Ichthyosis (ASIC) for their cooperation. This work was partially supported by Ramón Areces Foundation project (Rare Diseases 2013-056); by Spanish Instituto de Salud Carlos III (ISCIII) (INT15/00070, INT16/00154, INT17/00133) and by Xunta de Galicia (IN607B). UE was supported by a predoctoral fellowship from Xunta de Galicia. The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize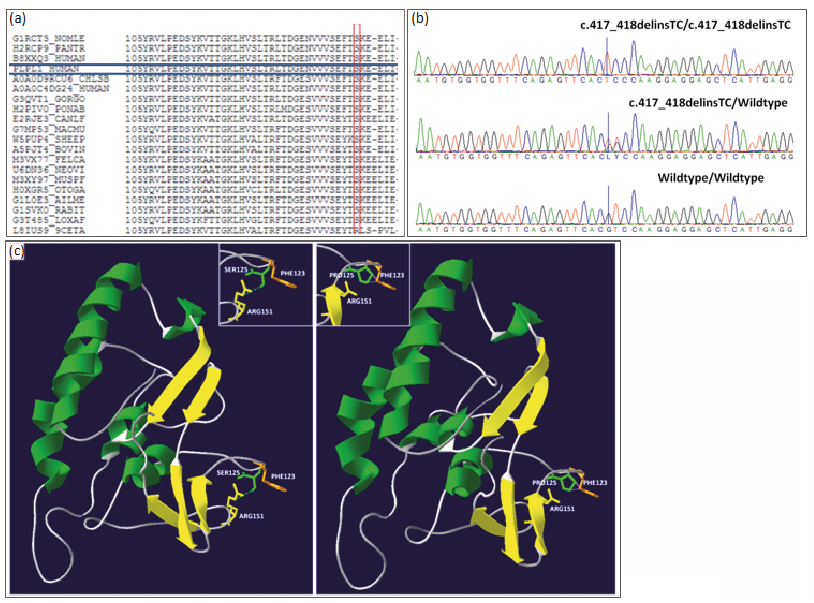 Click to show fullsize
Click to show fullsize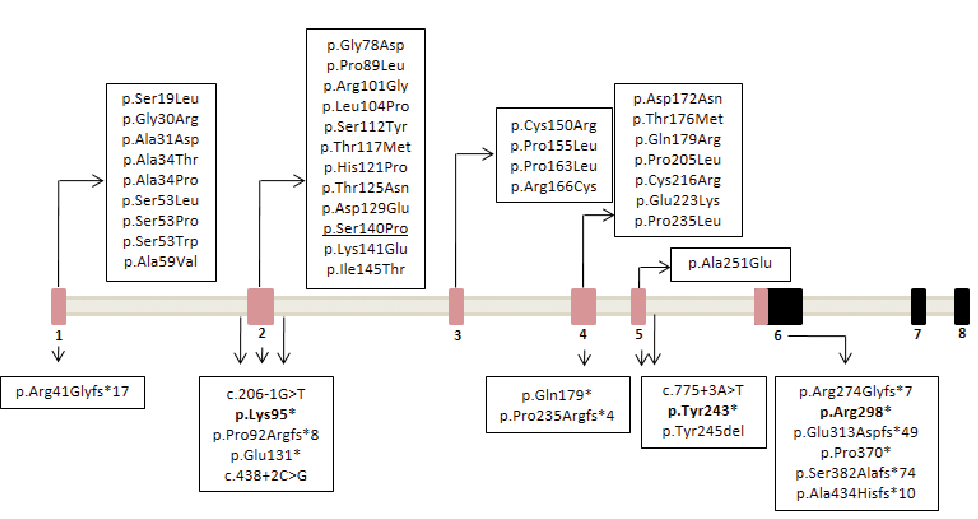 Click to show fullsize
Click to show fullsize