


1Paediatric Rheumatology Unit, 2Paediatric Ophthalmology Unit, Department of Woman and Child Health, 3Ophthalmology Department, and 4Neuroradiology Department, University of Padova, IT-35128 Padova, Italy. E-mail: giorgia.martini@aopd.veneto.it
Accepted Jun 13, 2019; E-published Jun 14, 2019
Juvenile localized scleroderma (JLS) comprises a group of autoimmune fibrosing conditions involving the skin and subcutaneous tissues following an initial inflammatory reaction. Linear scleroderma (LiS) is the most common subtype, characterized by one or more linear streaks of fibrosis, which can involve the dermis, subcutaneous tissue, muscle and, sometimes, bone (1). Although JLS is not a fatal disease, affected children may develop severe functional sequelae, such as joint contractures, limb growth discrepancy and psychological problems due to cosmetic disfiguration. Approximately one-quarter of patients present extracutaneous complications (2–4). Therefore, prompt diagnosis and treatment are necessary in order to reduce disease activity and halt tissue damage. We report here a girl who presented acquired strabismus as an unusual complication of LiS affecting the face.
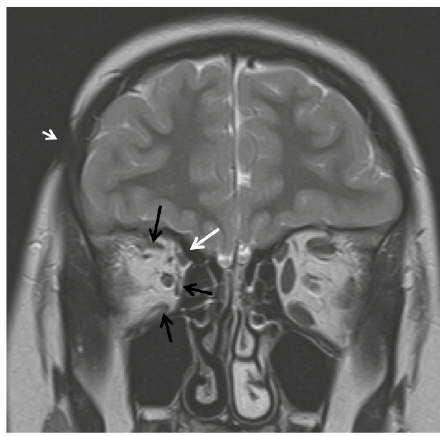
A 3-year-old girl developed a hyperchromic lesion with an ivory-like centre on the lateral aspect of her right eyebrow. The lesion subsequently extended to the temporal area, and 2 years later she presented intermittent exotropia. Cerebral computed tomography (CT) scan was normal and no treatment was advised. After 4 years she was referred to our unit and finally, at 7 years of age, a diagnosis of LiS of the face was made. On examination she presented right exotropia and hypotropia due to impairment of superior, medial and inferior rectus and superior oblique muscles. A linear violaceous lesion with moderate subcutaneous tissue loss and adnexa atrophy was present on the temporal area with small ivory-like centre. Cerebral magnetic resonance imaging (MRI) was performed and confirmed focal atrophy of subcutaneous fat tissue on the temporal area, compatible with scleroderma lesion, thinness and atrophy of superior, medial and inferior rectus and superior oblique muscles, but no cerebral abnormalities (Fig. 1). Treatment with methotrexate was commenced, and was continued for 2 years, in combination with prednisone for the first 3 months. A definite improvement in the skin lesion, with disappearance of inflammation and softening with hyperpigmentation, was observed with this treatment, while impairment of the extraocular muscles remained the same (Fig. 2).
Fig. 1. Coronal T2-weighted magnetic resonance imaging (MRI) image showing focal subcutaneous tissue loss in the right fronto-parietal region (arrowhead) and atrophy of the extraocular muscles of right orbit: superior, medial and inferior rectus (black arrows) and superior oblique (white arrow).
Fig. 2. (A) Skin lesion on the temporal area with hyperpigmentation and subcutaneous tissue atrophy (black arrow). (B) Right eye exotropia with elevation impairment. (C) Hyperfunction of superior oblique muscle causing elevation and adduction of the left eye. (D) Impairment of medial rectus muscle of the right eye. Written permission from the mother is given to publish these photos.
LiS is a rare disease, with an estimated annual incidence of 0.4–2.7 per 100,000 individuals. The overall incidence of JLS is not known, but there is evidence that the majority of cases occur during childhood (mean age at onset 7.9 years) (5, 6).
LiS is the most common form of JLS and, when occurring on the head, is referred as “en coup the sabre” (ECDS) due to the resemblance of the skin lesions to the stroke of a sabre (1). While it is believed that most patients will enter spontaneous remission after 3–5 years of disease activity, those with more extensive tissue involvement, such as in LiS, have a considerable risk of a more severe course (6). In fact, LiS tends to involve not only skin, but also subcutaneous tissue, muscles and bones, resulting in functional disabilities particularly when limbs are affected. In ECDS, involvement of underlying structures may cause facial deformities and neurological manifestations, and indeed cosmetic changes can markedly affect patients’ quality of life (7, 8).
In a large study of 750 patients with JLS, 22.4% presented extracutaneous involvement, such as articular, neurological, vascular, ocular, gastrointestinal, respiratory, cardiac, and renal manifestations. In more than one-quarter of these children, articular, neurological, and ocular involvements were unrelated to the site of skin lesions (4).
Ocular involvement in JLS is rare (3.2%), mostly associated with linear subtype, and generally related to extension of the lesions to annexes, resulting in abnormalities of eyelashes, eyebrows and eyelids, xerophthalmia and enophthalmus; more rarely inflammatory manifestations, such as episcleritis and anterior uveitis, as well as glaucoma and papilledema have been observed (9, 10). To the best of our knowledge, strabismus related to atrophy of extraocular muscles, as presented by this patient, has never been reported.
In general, the poor recognition of JLS clinical picture by primary care physicians accounts for a significant delay between disease onset and diagnosis and subsequent start of treatment and this probably negatively influences the course of disease, as described previously (2, 11–14).
In our patient, immunomodulatory treatment was started after 4 and 2 years from disease onset and from exotropia onset, respectively, probably when atrophic changes and irreversible damage in extraocular muscles had already established. Therefore, it is likely that such a long-time interval affected the possibility of functional recovery with treatment.
In conclusion, patients with LiS should be evaluated promptly as they can develop extracutaneous manifestations, such as in the current case, and clinicians should address proper investigations and prompt immunosuppressive treatment.


 Click to show fullsize
Click to show fullsize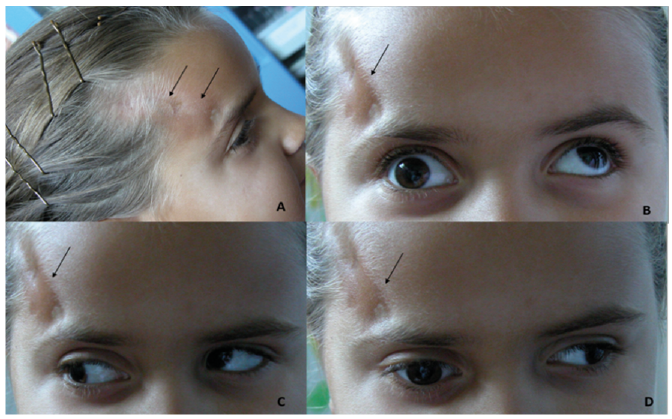 Click to show fullsize
Click to show fullsize