


1EpiFocus Ltd, London, UK, 2Galderma, La Tour-de-Peilz, Switzerland, and 3Cutaneous Lymphoma Foundation, Birmingham MI, USA
Most patients with mycosis fungoides are diagnosed with early-stage disease. However, prevalence of early-stage disease is unknown, and evidence of its burden is scarce. The aim of this study is to estimate the prevalence of early-stage mycosis fungoides, how long patients live with early-stage disease and to characterise these patients. Data were obtained from 4 key publications and from US cancer registries (Surveillance, Epidemiology and End Results Program; SEER). The derived incidence of early-stage mycosis fungoides was 0.26/100,000 (UK), 0.29/100,000 (US) and 0.38/100,000 (US-SEER) and the prevalence was 4.8/100,000 (UK), 5.2/100,000 (US) and 6.6/100,000 (US-SEER). Early-stage disease may last for 18 years. From SEER registries, 3,132 were diagnosed at early stage (mostly stage IA). Median age at diagnosis was 58 years. Compared with stage IA, the relative risk of death was 1.3 for stage IB and 3.5 for stage IIA. We confirm the rarity of early-stage mycosis fungoides, a differential prognosis and the potential for elevated burden of disease.
Key words: mycosis fungoides; stage; epidemiology; prevalence; prognosis.
Accepted Oct 28, 2019; E-published Oct 30, 2019
Acta Derm Venereol 2020; 100: XX–XX.
Corr: Andrew Maguire, Epidemiology, EpiFocus Ltd, London E11 2RJ, UK. E-mail: andy.maguire@epifocus.net
Most patients with mycosis fungoides are diagnosed with early-stage disease: European Organisation for Research and Treatment of Cancer stages IA, IB and IIA. However, prevalence of early-stage disease is unknown. Using results from key publications and US cancer registry data we estimated prevalence of 4.8/100,000 (UK), 5.2–6.6/100,000 (US) and that early-stage disease may last for 18 years. The cancer registries provided 3,132 early-stage mycosis fungoides patients. Compared with stage IA, the age and sex-adjusted relative risk of death was 1.3 for stage IB and 3.5 for stage IIA. This confirms the rarity of early-stage mycosis fungoides and a differential prognosis in early-stage.
Mycosis fungoides (MF) is the most frequent type of cutaneous T-cell lymphomas (CTCL), and most patients are diagnosed with early-stage disease (1–5). However, prevalence of early-stage disease is unknown and evidence of its burden and progression to advanced-stage disease is scarce.
In 2005, a consensus on CTCL classification was published. This harmonised the European Organisation for Research and Treatment of cancer (EORTC) and the WHO criteria and included MF (6). Despite the improved clarity in the identification of MF, there is evidence that diagnosis may occur approximately 3 years since onset of symptoms although this may be substantially higher (2, 5). MF was described as the most common CTCL typically affecting older patients with median age at diagnosis between 55 and 60 years old and affecting more men than women (1.6–2.0:1) (6). Progression from early to advanced-stage disease is typically slow (6). A refinement of the disease-stage classification was introduced in 2007 for MF, as well as for Sézary syndrome and has since been shown to correlate well with prognosis (1, 7). As well as considering the Tumour-Node-Metastasis (TNM) classification the staging also includes blood involvement (TNMB) (7). This 9-level classification divides the disease into early-stage (3 levels) and advanced, or tumour-stage, disease (4, 7, 8). Early-stage includes stages IA, IB and IIA. Stages IA and IB differ in the extent of lesions – the former with less than 10% of skin affected (7). Stage IIA includes some lymph node involvement (7). The 5-year disease-specific survival for these 3 groups varied from 98% (IA) to 89% (IB, IIA) (1). Thus, whilst considered “indolent” there is evidence of variability in the outcomes within early-stage disease (1, 2, 6). Furthermore, uncertainty on the risk of progression to advanced-stage disease is a concern for patients diagnosed at early-stage, with depression and anxiety being related to MF (9–11).
Currently, there is no cure for MF (12). Therapeutic skin-directed treatment of symptoms is recommended for early-stage disease, including topical corticosteroids and phototherapy (7, 13). Whilst both therapeutic options have been shown to be effective for many patients, there remains issues of toxicity related to long-term use of topical corticosteroids, and phototherapy. PUVA can increase skin cancer risk and hair loss as well as being time-consuming and inconvenient (14). As treatment is essentially palliative, the maintenance of quality of life (QoL) is the focus of treatment strategy (7, 12). Historically there has been no specific QoL instrument that can capture the experience of patients with MF until the recent development and validation of a new instrument, the MF/Sezary syndrome-CTCL QoL (15). It comprises 12 items. One of the items records whether the patient is worried that their disease may worsen for which the score in the sample used to develop the instrument was 2.74 (score ranges from 1 “best” to 5 “worst”) (15). This highlighted that many patients were worried about disease progression; the overall impact on quality of life was not reported. A different study, however, looked at the impact on QoL of skin diseases (16). Whilst MF was not examined, there was a high impact on depression and anxiety of chronic skin conditions and a 4-fold increase associated with pruritus, which is a typical symptom of MF. This highlights the high potential impact of symptoms associated with MF on the patients’ QoL.
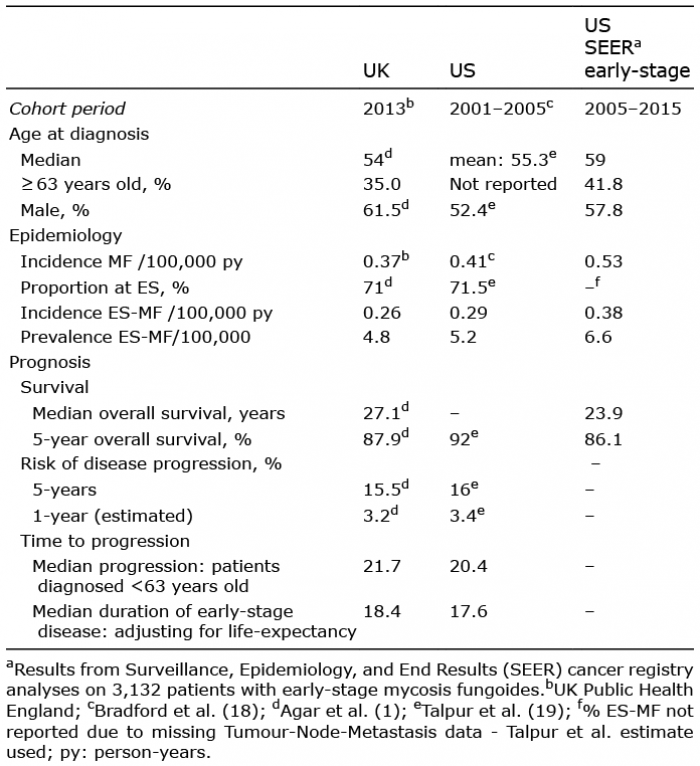
Prevalence is key to quantifying disease burden. If prevalence is unavailable it can be estimated by considering the incidence and the duration of the disease (17). Overall survival of MF, however, is not an appropriate measure of duration of early-stage MF as it does not contemplate the progression to advanced-stage disease. Indeed, the time a patient may expect to live with early-stage MF is also fundamental to understanding disease burden. Fortunately, the estimation in the UK and US of prevalence and the duration of early-stage MF are feasible from key publications from those countries. In the US, Bradford et al. (18) in 2009 provided the incidence rates of CTCL types including MF for the period 2001 to 2005. Also, in the US, Talpur et al. (19) provided the proportion of patients that are diagnosed at early-stage (71.5%) and described their prognosis in terms of progression-free survival for early-stage disease. In the UK, the incidence of MF was obtained from results provided by a report by Public Health England (20) whilst Agar et al. (1) examined the outcomes of MF and specifically provided the 5-year risk of progression by disease-stage. Agar et al. (1) provided a breakdown of MF by stage with 1,061 patients in early-stage categories.
In addition to US published data, the US National Cancer Institute provides cancer registry data on MF in its Surveillance, Epidemiology and End Results Program (SEER) (21) rendering bespoke analyses of these patients possible. However, the limitation of the SEER data is that progression to advanced-stage disease is not recorded.
Understanding how many patients live with early-stage MF and for how long underpins disease burden and unmet medical need. Early-stage disease will require long-term medical care, there is evidence of anxiety and, potentially, high impact on the patient’s QoL especially related to pruritic symptoms. Therefore, the aim of this study is to estimate, for the first time, the prevalence of early-stage MF and for how long patients live with early-stage disease. We also aim to characterise patients with early-stage disease and examine the potential differential prognosis between the 3 stages within early-stage disease.
Two approaches were used: a synthesis of published results and analyses of the SEER cancer registry data. Publications were obtained from PubMed using a broad search for epidemiological studies of MF or CTCL; this returned 616 studies published in the last 5 years and 462 review papers. These were screened and additional references mentioned in the papers were checked.
Disease progression
To estimate the progression rate from early to advanced-stage disease, we examined the main publications from the US and UK. From these, the annual rate was converted from the 5-year disease-progression risk as reported by Agar et al. (1) (UK) and Talpur et al. (19) (US). For the US, the 5-year rate was estimated from a Kaplan-Meier curve (19). The UK study provided tabulated 5-year rates for each of the 3 classes of early stage disease (IA, IB, IIA) and were thus weighted to estimate the overall rate for early-stage disease (1). The subsequent 5-year estimates were converted to annual rates, accounting for the cumulative nature of risk over the 5-year period. From these rates, the median duration of early-stage disease was estimated by assuming that the time to progression follows a typical survival curve as defined by an exponential decay function (17). Lastly, the estimate was adjusted by accounting for life-expectancy in those patients for whom the median time to progression was greater than their remaining life-expectancy.
Incidence and prevalence of early-stage disease
The proportion of new cases diagnosed with early-stage disease was applied to the overall incidence rates of MF. Both this proportion and the incidence rate were available for the UK and the US. Prevalence was then estimated by multiplying these incidence rates by the median duration of early-stage disease (17).
US Surveillance, Epidemiology and End Results Program analysis
The SEER data used for this study was comprised of patients from 18 cancer registries. These provide data on patients since 2000 and cover 26.4% of the US population (86 million). The SEER*stat software (version 8.3.5) was used to generate incidence rates as this incorporates appropriate denominators (22). Data on all patients diagnosed with MF since 2000 were then extracted from the SEER databases using SEER*stat. STATA (version 15.1) was used to generate descriptive analyses, graphs and to perform survival analyses (23). Cancer stage using the American Joint Committee on Cancer “AJCC” TNM 6th edition classification was extracted (24). This classification has been applied to patients diagnosed since 2004 included in the registries comprising the SEER program. The EORTC algorithm was then applied to those patients with valid TNM values diagnosed in 2005 or later to calculate the proportion diagnosed in each of the three early stages. This does not include the blood status of the patient as this classification was not available. The age-distribution was compared between the early and advanced-stage cases. Overall survival over the follow-up period (2005 to 2015) was examined, using life-tables to calculate the 5-year survival probabilities. Kaplan-Meier and Cox proportional hazards regression were applied to examine the impact of early-cancer stage, age and sex on overall survival (17). Lastly, as progression rates are unavailable in SEER, prevalence of early-stage disease was calculated by applying the proportion diagnosed at early-stage by Talpur et al. (19) and the median duration of early-stage disease as estimated in this paper.
Results were synthesised from 4 key publications (1, 18, 19, 20). In the US, Bradford et al. (18) provided an incidence rate of MF of 0.41/100,000 person-years and Talpur et al. gave the proportion diagnosed with early-stage disease as 71.5% (19) and also provided data on disease progression. Similarly, in the UK, the incidence rate of MF was obtained from Public Health England’s 2016 report; 0.42/100,000 person-years for men, 0.29 for women giving an overall incidence of 0.37 after accounting for the reported male-to-female ratio of 1.5:1.0 (20). Also, in the UK, Agar et al. reported that 71% were diagnosed with early-stage disease and reported results on disease progression (1). No other relevant publications were identified that can provide incidence and the proportion of early-stage disease for the same country as evidenced by the citations in recent reviews (2, 7).
Progression and duration of early-stage disease
In order to estimate the progression rate from early-stage to advanced-stage disease in the US, the approximate risk at 5 years since diagnosis of 16% was obtained from the Kaplan-Meier curves progression-free survival provided by Talpur et al. (19). This corresponded to an annual rate of 3.4% (Table I). For the UK, Agar et al reported the risk of disease progression at 5 years which ranged from 8% for stage IA to 21% for stage IB (1). After weighting in accordance with the relative frequency of stage occurrence, the 5-year risk of disease progression was 15.5%, corresponding to an annual rate of 3.2%. The UK and US progression rates, though derived from entirely independent sources, were therefore similar.
Table I. Epidemiology and duration of early-stage (ES) mycosis fungoides (MF)
These annual rates imply that the median time to progression would be 21.7 years for the UK and 20.4 years for the US. However, these do not account for life-expectancy. For both the UK and the US, life-expectancy is exceeded by the median time to progression for patients diagnosed at ages of 63 and above (25, 26). Given the age-distribution of patients diagnosed at 63 and above in the UK, the life-expectancy for this group was estimated to be 12.2 years. Thus, applying a time to progression of 21.7 years to patients younger than 63 years old and 12.2 years to those diagnosed at 63 and older, gives an expected median duration of early-stage disease for these patients of 18.4 years (Table I). For the US, the age-distribution was inferred from the SEER analyses as there was no information in the paper by Talpur et al. (19). The life-expectancy of people aged 63 or older in the US was 13.1 years (Table II) and thus the corresponding median duration of early-stage disease was 17.6 years.
Table II. Characteristics of patients diagnosed with mycosis fungoides in US Surveillance, Epidemiology, and End Results (SEER) cancer registries between 2005 and 2015
Incidence and prevalence
Incidence of MF was 0.37/100,000 person-years in the UK, whilst in the US it was 0.41 (18, 20). Analysis of the SEER registries for the period 2005 to 2015 gave a slightly higher incidence of 0.53/100,000 over this period. Applying the proportion of patients diagnosed at early-stage (UK: 71%; US 71.5%) gave corresponding incidence rates for early-stage MF of 0.26/100,000 and 0.29/100,000 in the UK and US, respectively (Table I). The higher incidence of MF as described for the US using current SEER incidence rates corresponded to an early-stage incidence rate of 0.38/100,000.
The product of these incidence rates with the country-specific duration of early-stage disease gave a prevalence for the UK of 4.8/100,000 people. In the US, the prevalence estimates varied from 5.2 to 6.6/100,000 people (Table I).
Age, stage and overall survival
There were 4,964 patients diagnosed with MF between 2005 and 2015 and included in the 18 cancer registries contributing to SEER (Table II). A total of 3,132 of these patients were categorised with early-stage disease when applying the EORTC algorithm. As 24% of patients had missing data for stage the proportion of patients diagnosed at early-stage compared with advanced is not reported.
Median age at which early-stage disease is diagnosed was 58 years old and a quarter of patients were diagnosed aged 69 or older (Table II). Patients diagnosed with early-stage disease were approximately 4 years younger than those diagnosed with advanced-stage MF. Among patients with early-stage disease the most frequent stage was IA (78.6%).
Overall 5-year survival rate was 86% for early-stage disease, and this decreased by stage from 87% for IA, 83% for IB to 58% for IIA (Table III). This is illustrated by the Kaplan-Meier curves of survival by the stages within early-stage disease (Fig. 1). Follow-up was restricted to 5 years due to low numbers in stage IIA.
The pattern observed in the survival curves is confirmed by Cox proportional hazards regression. The age- and sex-adjusted hazard ratio of 1.30 (95% confidence interval (CI) 1.01–1.67) indicated that the risk of death increased by around a third for those diagnosed with stage IB compared with IA (Table III). For those diagnosed with stage IIA there was a 3-fold increase in mortality compared with stage IA (hazard ratio 3.47; 95% CI 2.29–5.27).
Table III. Overall survival from diagnosis of 3,132 patients with early-stage mycosis fungoides (Surveillance, Epidemiology, and End Results (SEER) registries 2005–2015)
Fig. 1. Kaplan Meier curves of overall survival of patients with early-stage mycosis fungoides by disease stage.
This study provides the first estimate of the prevalence of early-stage MF with estimates ranging from 4.8 to 6.6/100,000 people. Furthermore, after accounting for life-expectancy, we expect that patients will live with early-stage disease for approximately 18 years. Burden of this disease can therefore be quantified by further consideration of the humanistic and health economic impact of early-stage MF.
The estimated duration of early-stage disease was similar in the UK and the US which was due to the similarity of the estimated annual disease progression derived from independent studies in each country (UK: 3.2%; US: 3.4%). However, there was notable variability in the prevalence estimates. This was due to differences in the incidence rates of MF. Since 2005 there have been changes in the definition of MF and potentially an increase in the awareness of this disease (7, 18). We used SEER data for the period 2005 to 2015 and the rates are therefore likely to reflect more accurately the current reporting habits for MF in the US than the previously published incidence rate. It is not clear, however, why the incidence rate in 2013 for the UK was lower. It may reflect differences between countries in reporting and recording potentially due to the rarity of the disease; geographical variation of CTCL has been suggested elsewhere (27). Furthermore, the issue of diagnostic accuracy remains an issue despite the EORTC guidelines (28). The potential for heterogeneity of the cases of MF may lead to differences in prognosis (27). Further research on the risk factors for progression is warranted and may provide insight into the composition of the cases.
While these results confirm the rarity of this disease, they also indicate the potential for elevated burden of disease due to the extremely long period for which patients need medical management. The burden is likely to increase as current treatment options may not provide a long-term, effective, solution for the management of symptoms (12). Effective and long-lasting management of symptoms is required due to the evidence that pruritic conditions can have a high impact on the patient’s QoL (9, 16). Ideally, new therapies would have the capacity to modify disease progression especially as prognostic uncertainty is a source of anxiety for patients with this cancer (9); the 2018 global patients survey on lymphomas highlighted the high levels of anxiety, depression and isolation suffered by patients with cutaneous lymphomas (9). Therefore, research into the impact of early-stage MF on QoL is required and may now be facilitated by the recent publication of a specific instrument for assessing QoL for MF and Sezary syndrome (15).
Evidence on the economic burden of early-stage MF has been sparse and is hampered by the difficulty to classify disease-stage in healthcare databases. However, recent analyses by Tsang et al (29) of resource utilisation and costs of MF inferred disease stage according to the use of systemic therapy. The main drivers of cost for “mild to moderate” cases (i.e., patients who did not use systemic treatment) were inpatient admissions and outpatient visits. However, whilst for these patients the mean total annual costs were USD28,903 there was very high variability, and hence uncertainty, as described by a standard deviation of USD82,847.
The SEER registries provided a large cohort of patients with MF for whom it was possible to infer their stage in accordance with the EORTC although a quarter of patients did not have their TNM stage recorded. The age of patients with missing TNM values was similar to those with advanced-stage disease. Hence, we may expect a bias which inflates the proportion of patients with early-stage disease; for this reason, these proportions are not reported. However, within early-stage disease the proportions of the specific disease stages (IA, IB, IIA) are likely to be accurate as will be the characteristics and survival experience.
Despite the focus on early-stage disease, we provided a comparison of the age at diagnosis between early and advanced-stage disease. Patients who were initially diagnosed with advanced-stage disease were 4 years older than those who were initially diagnosed with early-stage disease. Given the natural history of MF, this may imply a diagnostic delay and likely implies that these patients have lived longer with MF and have not benefitted from medical care. The role of the potential diagnostic delay warrants further research especially in the advent of new treatment options (12). The SEER analyses also provided a further breakdown of the early-stage categories. We observed that 79% of patients were diagnosed with stage IA. This contrasts with the UK result by Agar et al. (1) that this group comprises 41% (438/1061) among those diagnosed with early-stage disease.
Within early-stage patients, there was evidence that overall survival decreased with increasing stage. After adjusting for age and sex, we observed that the death rate increased by 30% for patients diagnosed at stage IB compared with stage IA whilst for those diagnosed with stage IIA the death rate was 3.5 times higher than stage IA. This confirms a previous report that the EORTC classification is correlated with prognosis (1).
In conclusion, this study quantifies and confirms the rarity of early-stage MF but it also highlights the potential for elevated burden of disease due to the extremely long period for which patients needs medical management. The observation that patients with early-stage disease are 4 years younger than those with advanced-stage disease may be evidence of diagnostic delay. It also confirms differential prognosis, after age and sex adjustment, within early-stage disease. Thus, effective and safe treatment options are required (12) and studies on the impact on the humanistic and economic burden of early-stage MF are warranted.
Galderma was the funding source and was involved in all stages of study conduct and analyses. Galderma also funded all costs associated with the development and the publishing of the present manuscript.
Conflicts of interest: AM was funded by Galderma to undertake the study (design, analyses and writing). JP, PR, RC and SG participated in the execution and review and are employees of Galderma. ST has no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize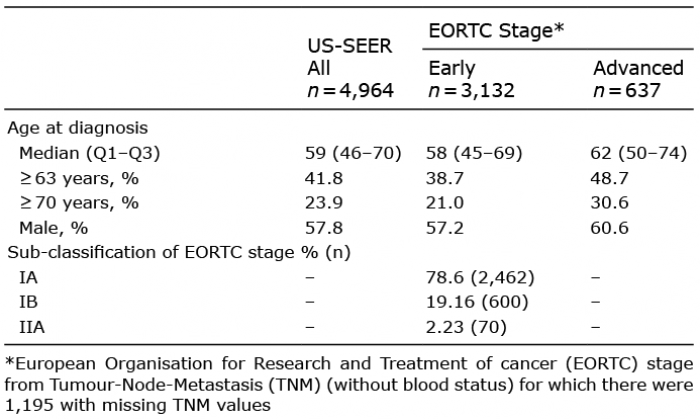 Click to show fullsize
Click to show fullsize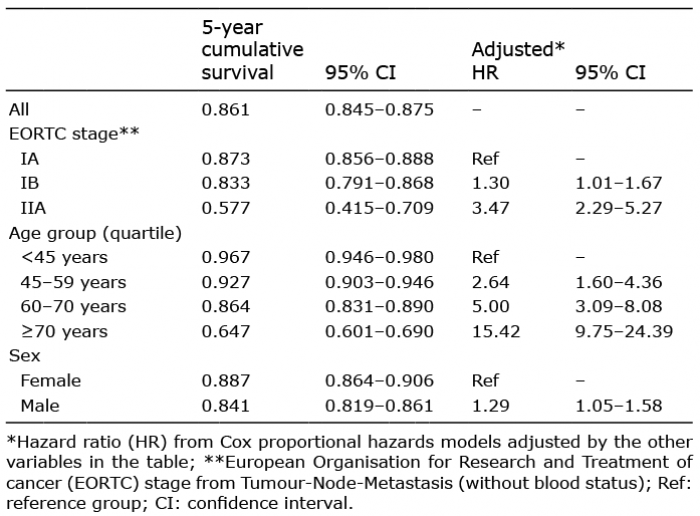 Click to show fullsize
Click to show fullsize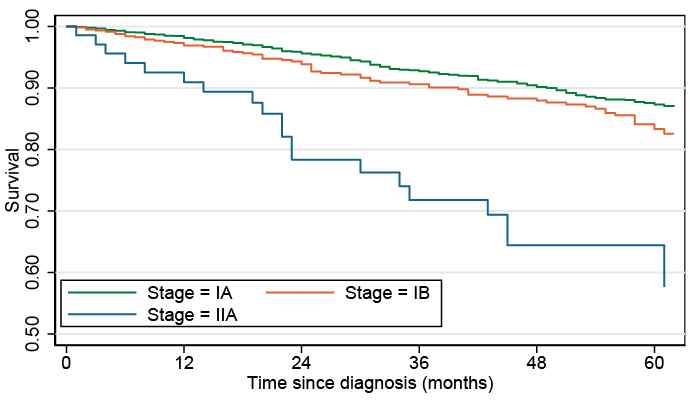 Click to show fullsize
Click to show fullsize