


1Institute of Genomic Medicine, 2Institute of Dermatology, 3Institute of General Pathology, and 4Internal Medicine, Gastroenterology and Hepatic Diseases Unit, Gastroenterological Area, Gastroenterological and Endocrino-Metabolical Sciences Department, F. Policlinico Gemelli IRCCS, Università Cattolica del Sacro Cuore, Largo F. Vito 1, IT-00168 Rome, Italy. *E-mail: simone.garcovich@unicatt.it
#These authors equally contributed to this work.
Accepted Nov 6, 2019; Epub ahead of print Nov 8, 2019
Acta Derm Venereol 2020; 100: adv00038
Combined immunodeficiency (CID) syndromes are rare conditions characterized by defective development and function of T-cell and B-cell immunity, determining an increased susceptibility to infections and a heterogeneous spectrum of muco-cutaneous manifestations (1). We report on a female patient presenting with a severe clinical phenotype of CID due to a novel, pathogenetic variant of the CARMIL2 gene. We further characterize the clinical spectrum of this recently described form of autosomal recessive CID with dual T and B-cell intrinsic deficiency (2).
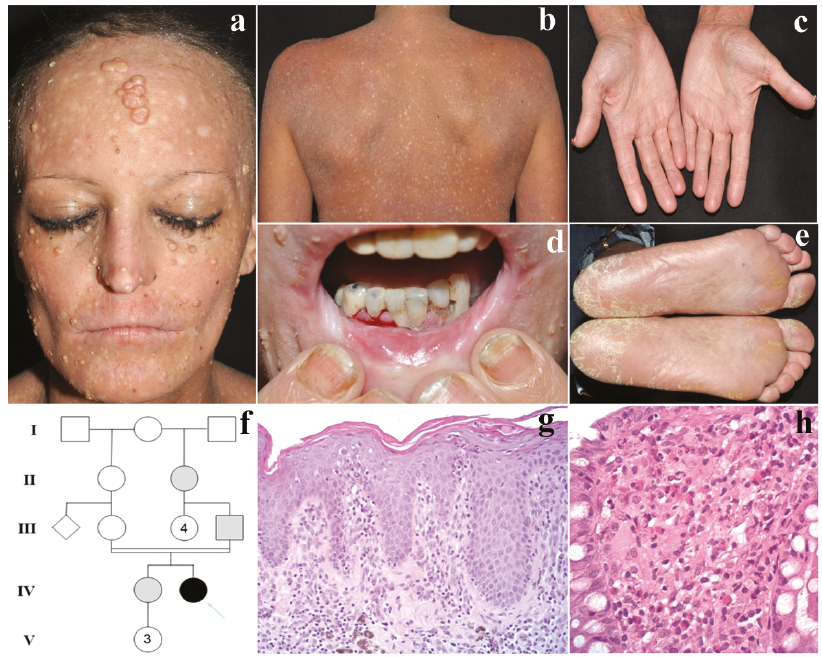
A 23-year-old woman was examined for diffuse atopic dermatitis-like lesions, which first appeared at the age of 18 months. She had consanguineous Italian parents (second-degree cousins), with a family history of psoriasis (see Fig. 1f for pedigree). Eczematous skin lesions evolved since childhood with a progressive-persistent course and with recurrent episodes of bacterial super-infection. From the age of 3 years, she underwent multiple hospitalizations, due to recurrent episodes of arthritis, bronchitis, gastrointestinal symptoms and severe iron-deficiency anaemia. At the age of 16 years, she developed pyorrhoea and chronic gingivitis, which progressively led to fibrosis of the oral mucosa and brittle teeth. In the following years, the patient developed extensive, giant molluscum contagiosum, refractory to conventional therapies (Fig. 1a–e), and chronic progressive dysphagia, due to acquired oesophageal stenosis, which was treated with periodic endoscopic dilations. Episodes of food allergy, with anaphylaxis and worsening of cutaneous and respiratory symptoms, were triggered by ingestion of certain foods (i.e. fish, eggs, milk/dairy products, strawberries).
Endoscopic, histological and laboratory work-up revealed psoriasiform dermatitis (Fig. 1g) and eosinophilic gastroenteritis (Fig. 1h), associated with chronic malabsorption-”leaky gut” syndrome, metabolic wasting and weight loss. Normal serum IgE levels and no abnormalities in blood leukocytes, immunoglobulin levels, and autoimmunity markers were detected. Whole exome sequencing allowed us to identify a homozygous novel nonsense variant in the CARMIL2 gene (OMIM* 610859) (NM_001013838.1, c.1109C>A, p.(S370*)), which is considered causative for the observed phenotype. In addition, real-time PCR and sequencing of CARMIL2 transcript on RNA extracted from peripheral blood white cells showed that the mutated allele was exclusively expressed, at similar levels to those of 2 unrelated controls. Immunophenotyping on peripheral blood T and B cells is described in Fig. S1. Similar to previously reported results (2), deficiency of human CARMIL2 did not interfere with CD4, CD8 and CD19 cells; although the percentage of cytotoxic T cells appeared higher than expected, the CD4/CD8 ratio was normal. Detailed analysis of T and B subpopulations compared with expected reference ranges (3) revealed, in this case, low counts of Treg in the peripheral blood, predominantly comprised of inducible and suppressor Treg, similar to that reported in other human patients and in mice with the same deficiency. On the other hand, contrary to other reports, the amounts of Th1 and Th17 subsets were in the normal range. B cells analysis showed low counts of memory B cells compartments (CD27+), balanced by an amount of naïve B cells and plasmablasts higher than reported in the literature (Fig. S1C) (2). Conservative therapeutic management of organ-specific manifestations included a combination of surgical removal and topical trichloroacetic acid for recurrent, giant molluscum contagiosum, as well as a proton-pump inhibitor, a “targeted elimination” diet, micronutrient supplementation and low-dose budesonide for gastrointestinal disease.
Fig. 1. Patient’s clinical features and pedigree. (a) Giant molluscum contagiosum involving facial skin. (b) Generalized chronic dermatitis with erythroderma. (c) Eczematous palmar skin. (d) Pyorrhoea, gingivitis, cheilitis with lip fissures. (e) Mild, plantar hyperkeratosis. (f) Pedigree of the studied family: the patient is indicated by an arrow and represented by a black circle, while grey symbols represent subjects with a very mild form of psoriasis. (g) Histology of lesional skin, showing a psoriasis-like chronic dermatitis pattern with parakeratosis. (h) Histology of colonic biopsy, displaying lymphomonocytic and granulocytic infiltrate with numerous eosinophils. Hematoxylin and eosin: g: × 200, h: × 400. Permission is given to publish these photos.
CARMIL2 (previously known as RLTPR) encodes capping protein regulator and myosin 1 linker 2, which plays an important role in T-cell dependent immunity. Interestingly, the gene was initially identified in psoriatic skin, where it is significantly down-regulated (4). However, recent studies have shown that CARMIL2 is essential for CD28 co-stimulation and the concomitant activation of NF-?B pathway in human T cells (5–7). To date, primary CID due to complete functional CARMIL2 deficiency, resulting from biallelic variants, has been described in 30 patients (reported in Table SI) (2, 8–13).
Affected patients primarily show a consistent reduction of Treg cells in peripheral blood, but also of other CD4+ T cells, including Th1, Th17, and Tfh cell subsets. Deficient function of the CD28-CARMIL2-CARD11 pathway leads to impaired development of Th1 and Th17 lymphocytes and Th2-biased immune imbalance, with consequent risk of infections and allergic hypersensitivity reactions of the epithelial barrier-organs.
The clinical features of immune dysregulation mainly affect the skin and oral, digestive and respiratory mucosae, with an increased risk of viral, bacterial and fungal infections (see Table SI). Muco-cutaneous manifestations include atopiform or psoriasiform dermatitis, photodermatitis, warts or molluscum contagiosum, dermatophytosis, chronic mucocutaneous candidiasis, subcutaneous abscesses, chronic stomatitis/cheilitis and gingivitis. Also, very rare Epstein-Barr virus (EBV)-related smooth muscle tumours (SMT) have been reported, due to impaired antiviral immunity (8, 13). Non-infectious, autoimmune or allergic manifestations are variably reported in patients with CID (1). In CARMIL2-deficiency, allergic and hypersensitivity disorders seem to be more prevalent than autoimmunity, due to depletion of Tregs and defective CD28 co-stimulation (6). Allergic asthma, rhinitis and food allergy are a distinctive feature of this condition, comprising also rare conditions, such as eosinophilic esophagitis and colitis, as observed in our case (14). Moreover, CARMIL2-deficiency has recently been reported in children with very early onset of inflammatory bowel disease and eosinophilic gastrointestinal disorders, even in the absence of overt immunodeficiency (12).
Even though no clear-cut genotype–phenotype correlations can be drawn, it appears that variants more likely causing a complete loss of protein function, with absent protein expression, are associated with a more severe phenotype compared with missense variants affecting the LRR domain. However, the nonsense variant we found in our patient seems not to trigger a nonsense-mediated decay (since the transcript is expressed in peripheral blood cells), suggesting that some amount of a truncated CARMIL2 protein is produced. Haematopoietic stem cell transplantation represents the only potentially curative treatment option for primary CIDs, but its indication should be decided based on the specific type of condition and the patient’s characteristics (15). In conclusion, our report provides further insights into the new nosological entity of CARMIL2-related CID, and highlights the extensive spectrum of muco-cutaneous manifestations. Dermatologists should be aware of the role of CARMIL2 deficiency, especially in patients presenting with persistent/disseminated viral infections and atypical “atopiform” or “psoriasiform” skin manifestations.


 Click to show fullsize
Click to show fullsize