


1Department of Dermatology and Allergy, Herlev and Gentofte Hospital, University of Copenhagen, DK-2900 Hellerup, and 2Department of Otolaryngology, Head and Neck Surgery and Audiology, Rigshospitalet, Copenhagen, Denmark. *E-mail: kristian.kofoed@regionh.dk
Accepted Jan 27, 2020; Epub ahead of print Jan 29, 2020
Acta Derm Venereol 2020; 100: adv00052
The term urticaria haemorrhagica profunda was first proposed by Wollenberg et al. (1) in 1997 to describe the unusual clinical presentation of urticarial vasculitis and subcutaneous haemorrhage. Urticarial vasculitis is a rare disease characterized by recurrent long-lasting urticarial lesions with histopathological evidence of leucocytoclastic vasculitis. The disease can be further characterized according to complement levels as either normo- or hypocomplementemic urticarial vasculitis, of which the latter tends to be more severe (2). Cutaneous haemorrhage without preceding trauma or underlying bleeding disorder is described in early childhood as acute haemorrhagic oedema of childhood. Like urticarial vasculitis, the histopathological finding is leucocytoclastic vasculitis, but clinically the disease presents as an ecchymotic rash with pronounced oedema, predominantly over the face, ears, and limbs (3). We present a case of hypocomplementemic urticarial vasculitis (HUV) and severe, spontaneous, subcutaneous haemorrhage in an adult patient.
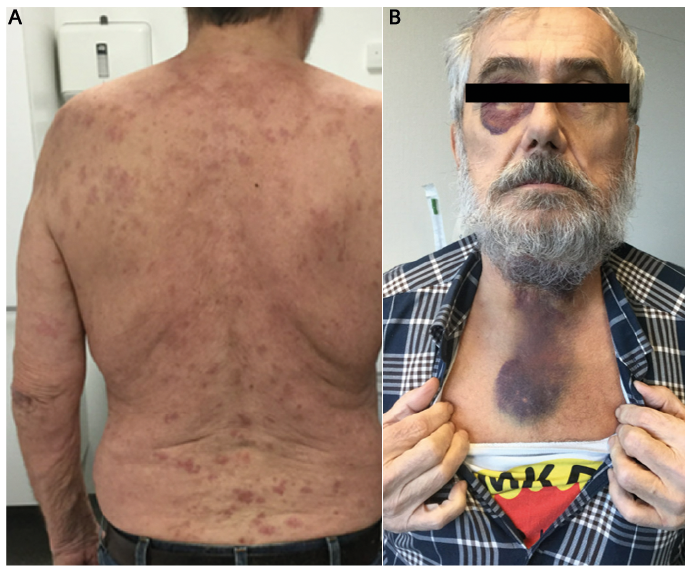
We report the case of a 75-year old man with a two-year history of recurrent long-lasting urticarial lesions, before an acute change of symptoms. He was otherwise healthy and took no medications. He described the urticarial lesions as painful rather than pruritic. They lasted > 24 h and healed spontaneously with residual post-inflammatory hyperpigmentation. Without any apparent triggers or prodromal symptoms, he experienced an acute flare of the usual urticarial lesions (Fig. 1A), accompanied by arthralgias, abdominal pain, oedema of the upper lip, and dysphagia. In the emergency room, he was suspected of having an acute allergic reaction with an evolving angioedema. Laryngoscopy showed haemorrhage in the fauces, the tongue and the laryngeal mucosa. He was treated with intravenous clemastine 2 mg once and methylprednisolone 80 mg daily for two days. Four days later, he developed massive, subcutaneous haemorrhagic lesions, in the periorbital region, and on the anterior neck (Fig. 1B). Histopathological examination of a skin biopsy from an urticarial lesion showed endothelial cell damage with a neutrophil-predominant perivascular infiltrate, leucocytoclasis, and erythrocyte extravasation. Amyloidosis was excluded on tongue biopsy.
Fig. 1. (A) Widespread urticarial lesions on the patient’s back. (B) Massive, subcutaneous haemorrhagic lesions, located to the periorbital region, and the anterior neck.
Blood samples showed consumption of complement with a complement C1q level of 0.24 mmol/l, (normal range 0.3–0.96 mmol/l), complement C3 level of 0.371 g/l (normal range 0.811–1.570 g/l), and complement C4 level of 0.022 g/l (normal range 0.129–0.392 g/l). Further, there was massively elevated creatine kinase level of 6,034 U/l (normal range 40–280 U/l), myoglobine level of 3,030 mmol/l (normal range 24–77 mmol/l), and elevated lactate dehydrogenase of 459 U/l (normal range 115–225 U/l). Complement C1q antibodies were negative. Anti-nuclear antibodies, and antibodies against double stranded DNA, P-DNA topoisomerase 1, Smith, beta-2-glycoprotein, cardiolipin, anti-Sjögren’s-syndrome-related antigen A and B, U1 ribonucleoprotein, and proteinase 3 were all negative. Serological tests for polymyositis included polymyositis anti-signal recognition particle antibodies, Ku antibodies, PL-7 antibodies, centromere antibodies, 52 kDa Ro protein antibodies, glycyl tRNA synthetase antibodies, MDA-5 antibodies, MI2 alpha antibodies, MORC3 antibodies, PL-12 antibodies, exosome component 10 antibodies, exosome complex RRP45 antibodies, histidine-tRNA-ligase antibodies, fibrillarin antibodies, SUMO activating enzyme subunit 1 antibodies, E3 ubiquitin protein ligase TRIM33 antibodies. All were negative. Hepatitis B, C, and E antibodies were negative. Creatinine was normal and there was no proteinuria. C-reactive protein and alanine transaminase were within normal limits. Haemoglobin was normal initially (8.8 mm/l) but during the course of the disease there was a slight drop to 7.9 mm/l with no other evidence of haematological disease. A positron emission tomography of the neck, thorax, and abdomen was normal.
The urticarial lesions and ecchymoses responded well to oral prednisolone 50 mg daily, tapered over 21 days. Creatine kinase and myoglobin levels responded to intensive fluid resuscitation. Two months later he experienced another spontaneous flare of urticarial lesions and similar massive subcutaneous haemorrhage and treatment with oral methotrexate 15 mg weekly was initiated. The treatment was well tolerated and at follow-up 3 months later, there had been no further episodes of urticarial vasculitis or haemorrhagic lesions.
Urticarial vasculitis is a leukocytoclastic vasculitis that is clinically characterized by longer lasting urticarial lesions. Although the causality is not clear, its association to a wide variety of systemic diseases has been reported, including hepatitis B and C (4), infectious mononucleosis (5), Lyme disease (6), haematological disorders and malignancies (4), Sjogren’s syndrome (7), and systemic lupus erythematosus (8).
The disease is categorized according to levels of complement as either normo- or hypocomplementemic. Both subtypes may be idiopathic, but the hypocomplementemic variant tend to have a more severe course and is often associated to systemic inflammatory disease (9). In particular, an association between HUV and systemic lupus erythematosus has frequently been reported (8, 10, 11).
In 2012, the American College of Rheumatology defined HUV as vasculitis accompanied by urticaria and hypocomplementemia affecting small vessels, and associated with anti-C1q antibodies (12). The relationship between idiopathic normocomplementemic urticarial vasculitis (NUV) and HUV is not well defined. However, several authors have suggested that NUV may progress into HUV (2, 4, 13, 14). Patients with mild disease should be regularly monitored to detect any signs or symptoms suggesting progression to a more severe subtype, as the course of the disease in this case exemplifies. Our patient suffered from recurrent urticarial vasculitis with no complications or associated disease for two years before the sudden change of symptoms.
Angioedema may be present in as many as 40% of patients with urticarial vasculitis, frequently involving the lips, tongue, periorbital tissue, and hands (15). Initially, the clinical picture may be interpreted as urticaria and allergic angioedema with acute endangered airway requiring immediate airway management. However, the facial manifestations in our patient were, predominantly haemorrhagic, not oedematous as classical angioedema.
To our knowledge, this is the third case of urticarial vasculitis accompanied by haemorrhagic lesions of the face and orolaryngeal mucosa in an adult patient ever described. The first case was described more than 100 years ago by Wills & Lond (16). In 1997, Wollenberg et al. (1) described the exact same symptoms in a 16 year old boy and another similar case was reported in 2003 (17). Histopathologically, the haemorrhagic lesions in all reported cases are similar to those of acute haemorrhagic oedema of childhood, and show signs of leucocytoclastic vasculitis. This severe HUV accompanied by haemorrhage could represent an adult equivalent of acute haemorr-hagic oedema of childhood. As angioedema is the deep dermal and subcutaneous equivalent of urticaria, there may be a pathophysiological relationship between superficial urticarial vasculitis and a deeper haemorrhagic vasculitis, as presented in our case. Wollenberg et al. (1) were the first to present this hypothesis and the authors suggested the term “urticaria hemorrhagica profunda” to describe this rare clinical picture.


 Click to show fullsize
Click to show fullsize