


1Department of Dermatology and Allergy, Allergie-Centrum-Charité, Charité – Universitätsmedizin Berlin, 2Autoinflammation Reference Center Charité (ARC2), Charité – Universitätsmedizin Berlin, 3Department of Pediatric Pneumology and Immunology, Center for Chronically Sick Children, Charité – Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt-Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany, and 4Department of Dermatology and Allergy Center, Kansai Medical University, Hirakata, Japan
#These authors contributed equally and should be considered as co-first authors.
Autoinflammatory diseases comprise a group of chronic disabling entities characterized by inflammation without the presence of infectious agents, auto-antibodies or antigen-specific T-cells. Many autoinflammatory diseases are caused by monogenic defects, which lead to disturbed immune signalling with release of proinflammatory mediators. In addition to interleukin-1β and interleukin-18, interferons play a key role in the pathophysiology of these disorders. Patients with autoinflammatory diseases show a broad variety of clinical symptoms, including skin involvement. Wheals, pustules and ulcerative lesions are the most common cutaneous findings observed. Knowledge of the clinical presentation of autoinflammatory diseases is crucial for establishing the diagnosis and guiding appropriate treatment. This review focuses on the dermatological findings in selected autoinflammatory disorders based on their distinct pathomechanisms.
Key words: autoinflammatory; genetics; interferon; interleukin-1.
Accepted Feb 12, 2020; Epub ahead of print Mar 9, 2020
Acta Derm Venereol 2020; 100: adv00091.
Corr: Karoline Krause, MD, Department of Dermatology and Allergy, Charité – Universitätsmedizin Berlin, Charitéplatz 1, DE-10117 Berlin, Germany. E-mail: karoline.krause@charite.de
Autoinflammatory diseases are rare disabling disorders characterized by excessive inflammation of the skin and inner organs. Many autoinflammatory diseases are caused by genetic defects, which subsequently result in disturbed immune signalling. In the skin, wheals, pustules and ulcerative lesions dominate. As autoinflammatory diseases are associated with a high burden and limited awareness, knowledge of their clinical presentation is crucial for establishing the diagnosis and guiding appropriate treatment.
Autoinflammatory diseases are a group of chronic disabling entities characterized by self-directed inflammation, which is mediated via disturbances in innate immune signalling pathways. The term “autoinflammatory” was established in the late 1990s to classify systemic diseases that lack high-titre autoantibodies and autoreactive T cells as known from autoimmune diseases (1). Within the last 2 decades, the spectrum of autoinflammatory diseases has grown rapidly. In addition to rare monogenic entities, it comprises a variety of multifactorial diseases with variable onset. Even for common disorders, such as gout, cardiovascular, metabolic and neurodegenerative diseases, autoinflammatory disease mechanisms have been claimed (2–5). Furthermore, the coexistence of both autoinflammatory and autoimmune features in several inflammatory disorders demonstrates the close link between the innate and adaptive immune signalling cascades (6).
As a joint disease pathomechanism, excessive cytokine secretion from innate immune cells (e.g. macrophages, monocytes) drives the inflammation in various organs. In particular, the accumulation of interleukin (IL)-1-associated cytokines, including IL-1β and IL-18, plays a crucial role in many diseases. In addition, increased amounts of interferons (IFN) have been recognized as the main inflammatory mediators in other conditions (7).
The clinical presentation of autoinflammatory diseases comprises recurrent fever attacks, musculoskeletal, gastrointestinal and neurological involvement. Also, the skin is affected in many of these disorders. Typical symptoms include urticarial, pustular and ulcerative lesions. This review focuses on the dermatological findings in selected autoinflammatory disorders based on their distinct pathomechanisms.
In 1984, the nucleotide sequence of IL-1 was identified, and decades of research revealed its importance as a central mediator of innate immunity and inflammation (8). The human IL-1 family consists of a total of 11 members with distinct biological functions (9). Among them, the proinflammatory cytokine IL-1 is the best-characterized member, composed of 2 individual forms, IL-1α and IL-1β. IL-1β is the predominant circulating isoform of IL-1 and initiates a cascade of activities in almost every tissue during host defence against pathogens and injuries. IL-1α and IL-1β exert their action through binding to a single ubiquitously expressed membrane-spanning receptor, known as IL-1 receptor type 1 (IL-1R1) (10). The binding of IL-1 to IL-1R1 mediates a conformational change that allows the co-receptor IL-1R accessory protein to bind. Hence, the trimeric complex triggers a signalling cascade, leading to the activation of NFκB. The naturally occurring IL-1 receptor antagonist (IL-1RA) competes with free IL-1, whereby interaction with its receptor is prevented (11).
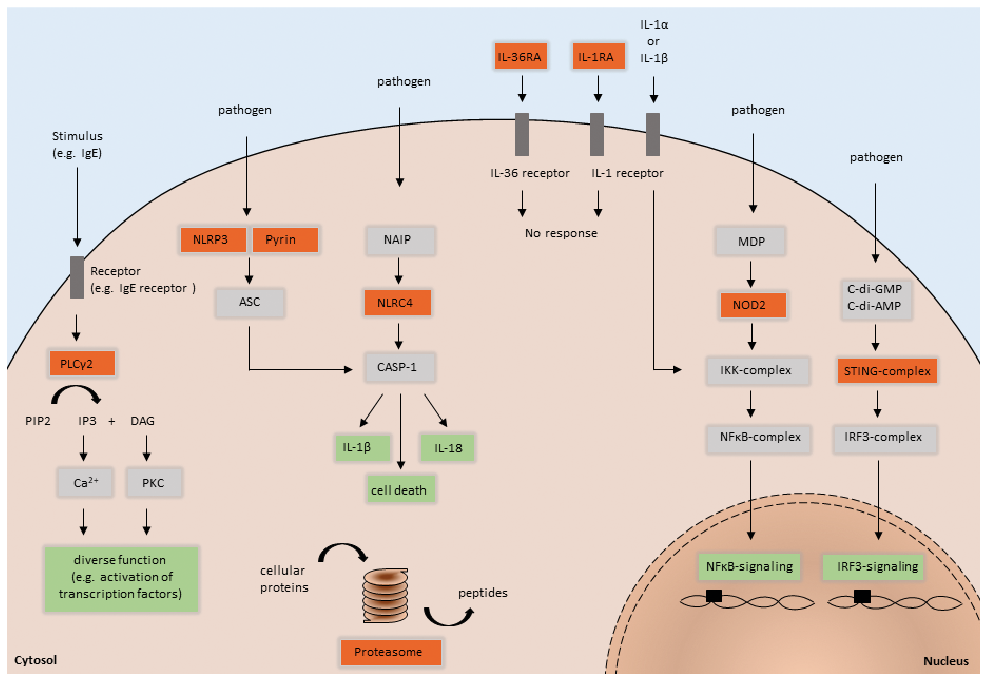
Inflammasomes are multimeric protein complexes and play a crucial role in the cleavage of pro-IL-1β. Cryopyrin, encoded by the NLRP3 gene, is a member of the NOD-like receptor family and is expressed by monocytes, granulocytes, T cells, chondrocytes, keratinocytes and mast cells (12). It is a protein that consists of 3 domains: an amino-terminal pyrin domain (PYD), a central nucleotide-binding and oligomerization domain (NACHT) and a C-terminal leucine-rich repeat (LRR) domain. The PYD is crucial for the assembly of the nucleotide-binding domain like receptor protein 3 (NLRP3) inflammasome, an intracellular macromolecular structure responsible for recognition of dangerous signals and important for host immune defence against pathogens (13, 14). In detail, the PYD of the cryopyrin interacts with the PYD of an adapter molecule, known as apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), and leads to the activation of the precursor protein pro-caspase-1. The activated caspase-1 contains a processing activity, whereby pro-IL-1β is cleaved to the mature active form (IL-1β). The synthesis of biologically inactive pro-IL-1β is mediated by NF-κB binding to the consensus binding site to transcribe the IL-1β gene (15, 16).
Cryopyrin-associated periodic syndrome
Cryopyrin-associated periodic syndrome (CAPS) is the prototype hereditary inflammasomopathy, with over 200 different underlying heterozygous gain-of-function mutations within the NLRP3 gene (INFEVERS database; https://infevers.umai-montpellier.fr/web/index.php, accessed November 2019).
These NLRP3 mutations, mainly concentrated in exon 3, constitutively activate cryopyrin, leading to increased conversion of pro-IL-1β into its active form with subsequent IL-1β hypersecretion (Fig. 1) (17, 18). CAPS consists of a group of 3 phenotypes: familial cold autoinflammatory syndrome (FCAS) as the mildest subform, Muckle-Wells syndrome (MWS) as the intermediate variant, and neonatal-onset multisystem inflammatory disease (NOMID) as the most severe phenotype (19, 20). Patients with FCAS present with cold-induced skin symptoms and musculoskeletal complaints. Patients with MWS show additional neurosensory hearing loss and may develop amyloidosis, whereas patients with NOMID have bone malformations and can develop severe neurological defects caused by aseptic meningitis (Table I). The physical complaints mainly start in early childhood or adolescence, but can also occur later in life due to rare cases of somatic mutations. The symptoms in CAPS are often accompanied by recurrent fever episodes and elevated levels of inflammatory markers, such as C-reactive protein (CRP), leukocytosis, serum amyloid (SAA) and S100 A8/9 or A12 (21). The crucial role of IL-1β in the pathogenesis of CAPS was proven by increased IL-1β secretion from leukocytes of patients with CAPS and highly effective anti-IL-1 treatment (22–26).
Fig. 1. Schematic representation of innate immune pathways and related pathomechanisms of the described autoinflammatory diseases. Red squares highlight the position of mutations associated with PLCG2-associated antibody deficiency and immune dysregulation (PLCγ2), autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (PLCγ2), cryopyrin-associated periodic syndrome (NLRP3), familial Mediterranean fever (Pyrin), nucleotide oligomerization domain (NOD)-like receptor family CARD domain-containing protein 4-inflammasomopathy (NLRC4), deficiency of interleukin-36 receptor antagonist (IL-36RA), deficiency of interleukin-1 receptor antagonist (IL-1RA), Blau syndrome (NOD2), STING-associated vasculopathy with onset in infancy (STING-complex), and chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature/proteasome-associated autoinflammatory syndrome (Proteasome).
Table I. Overview of affected genes, epidemiology, main clinical findings, skin symptoms and treatment options in selected autoinflammatory diseases grouped by their pathophysiological mechanisms of inflammation
Patients with CAPS present with an urticarial or maculo-papular rash, which is often symmetrically distributed on the trunk and/or extremities (Fig. 2). Skin lesions usually occur on a daily basis, last for up to 24 h and aggravate during the course of the day, with a peak in the evening (27). In patients with FCAS and those with MWS, the urticarial rash and systemic symptoms are triggered and exacerbated by cold air or evaporative cooling of the skin. In contrast, direct cold exposure does not induce the skin symptoms (27, 28). The skin lesions are rarely itchy, but can be accompanied by burning sensations and pain (27).
Based on its rarity, there is only limited data on the characteristics of skin inflammation in patients with CAPS. Skin histopathology is characterized by a neutrophil-rich dermal infiltrate (29–31). Accumulation of IL-1β and IL-6 after cold provocation testing was shown in lesional skin of patients with FCAS and dermal mast cells were identified as main producers of IL-1β (23, 32). In addition, IL-17-positive cells were observed in FCAS skin. These are believed to be stimulated by IL-1β, resulting in neutrophil recruitment and further production of IL-17 (33). The urticarial rash is thought to be mediated by NLRP3 inflammasome activation and consecutive IL-1β production of skin mast cells. IL-1β leads to vascular leakage und neutrophil accumulation as the pathological hallmark in neutrophilic urticaria.
Fig. 2. Urticarial rash on the right arm in a 77-year-old woman with cryopyrin-associated periodic syndrome.
Familial Mediterranean fever
Familial Mediterranean fever (FMF) is mostly an autosomal recessive disease caused by mutations within the MEFV gene, encoding a 781-amino acid pyrin/marenostrin protein (34, 35). Pyrin has a PYD and an N-terminal homotypic interaction domain, expressed by monocytes, granulocytes, dendritic cells and synovial fibroblasts (36, 37). To date, around 300 different mutations of the MEFV gene have been reported (INFEVERS database, accessed November 2019). The inflammation of FMF is mediated by ASC-dependent, NLRP3-independent production of IL-1β due to gain-of-function pyrin mutations (Fig. 1) (38).
The main clinical findings in patients with FMF comprise recurrent self-limiting attacks of fever and serositis as well as peritonitis, synovitis and pleuritis (Table I) (39). There is considerable inter-individual variability in the intensity and frequency of attacks. Between attacks, most patients with FMF are asymptomatic. In general, onset occurs within the first 2 decades and the disease becomes more severe during the course of life (40, 41). In untreated patients, amyloidosis can develop, with subsequent kidney failure (42, 43). Laboratory indicators are elevated acute-phase reactants, similar to those in patients with CAPS (see above) (44).
In up to 40% of patients with FMF, erysipelas-like skin lesions are reported. Those non-infectious lesions mostly affect the lower extremities and present as erythematous, painful infiltrated oedema (40, 45). Erysipelas-like lesions resolve spontaneously within several days and can be accompanied by fever and/or arthralgia (Fig. 3) (41). These skin lesions are typical for patients with FMF and do not occur in the context of other autoinflammatory disorders. Histopathologically, erysipelas-like lesions show dermal oedema and sparse perivascular infiltration of lymphocytes and neutrophils. Direct immunofluorescence revealed deposition of C3 in the small vessel wall of the superficial vascular plexus (46). Also, a strong association of FMF with polyarteriitis nodosa and Henoch-Schönlein purpura was reported (47–49). Less frequently, patients with FMF can present with other skin symptoms, such as purpuric exanthema and urticarial rash, diffuse palmoplantar erythema, Raynaud-like phenomena and erythema nodosum (50, 51). As a hypothesis, the co-occurrence of numerous immune-mediated disorders may be linked with inappropriately polarized T-cell responses in FMF, which enhances the occurrence of Th1- and Th17-driven diseases (52, 53).
Fig. 3. Patient with familial Mediterranean fever with erysipelas-like lesion of the left lower leg and accompanying arthritis of the left ankle joint.
Deficiency of interleukin-1 receptor antagonist
Deficiency of IL-1 receptor antagonist (DIRA) is an auto-somal recessive autoinflammatory disorder caused by a homozygous mutation in IL1RN, a gene that encodes IL-1RA, which inhibits the pro-inflammatory cytokines IL-1α and IL-1β (Fig. 1) (54). Several disease-causing mutations have been reported, including missense mutations, nonsense mutations and deletions (54–59). Due to these mutations, IL-1 signalling is increased leading to uncontrolled systemic inflammation.
Onset of symptoms occurs at birth or at the age of few weeks. Patients with DIRA present with multifocal osteomyelitis accompanied by severe bone inflammation and consecutive osteolytic changes and osteopenia, periostitis and pustulosis (Table I) (55). The disease is characterized by premature birth and failure to thrive, as well as respiratory distress. Abnormal laboratory findings include leukocytosis with elevated inflammatory markers and anaemia despite the absence of fever (54, 55).
Cutaneous findings range from the occurrence of disseminated small pustules to severe generalized pustulosis and may be accompanied by ichthyosis. They are mainly located on the trunk and the extremities (54, 55). In most patients with DIRA, the pustular dermatitis is associated with nail dystrophy, such as onychomadesis (60). As the nail matrix is integrated in the enthesis of the extensor tendons, bone inflammation may merge into enthesitis and nail involvement. Histopathologically, DIRA is characterized by epidermal acanthosis and hyperkeratosis (55). The lesional epidermis and dermis is infiltrated by extensive amounts of neutrophils that form subcorneal pustules (54, 55). The exact mechanisms of pustule formation in patients with DIRA is not known. Activation of proinflammatory cytokines including IL-8 may mediate the expansion of IL-17-producing T cells, leading to consequent cutaneous neutrophilic influx and pustule formation. In line with this, IL-17 expression is upregulated in DIRA compared with controls (54). In contrast to urticarial neutrophilic dermatoses, autoinflammatory pustular disorders are characterized by epidermal involvement contributing to skin inflammation. Further investigations are necessary to clarify the role of keratinocytes and antimicrobial peptides, such as LL-37/cathelicidin, to better distinguish disease pathomechanisms.
Deficiency of interleukin-36 receptor antagonist
Analogously to DIRA, deficiency of IL-36 receptor antagonist (DITRA) is caused by a recessive homozygous or compound heterozygous mutation in the IL-36RA gene, resulting in deficiency of the IL-36 receptor antagonist (Fig. 1) (61). Consequently, pro-inflammatory cellular signals via IL-36 are enhanced, leading to systemic inflammation and generalized pustulosis. Patients with DITRA present with attacks of fever, elevated inflammatory marker CRP and leukocytosis with neutrophilia. In contrast to DIRA, there is no bone inflammation or involvement of inner organs in patients with DITRA (Table I). This can be explained by the fact that IL-36RA is physiologically mainly expressed in the skin and absent in bones or solid organs. Hallmarks of skin symptoms are flares of generalized pustulosis, as observed in patients with DIRA (61) (Fig. 4). Mutations in the IL36RA gene can also cause acrodermatitis continua of Hallopeau, a sterile pustular eruption, mainly acrally located with subsequent affection of the nails (62). DITRA is often named as a monogenic form of pustular psoriasis. In addition to IL36RA, mutations in CARD14 and AP1S3 have been identified in patients with generalized pustular psoriasis. All these mutations lead to enhanced IL-36 signalling with subsequent systemic and skin inflammation (63–65). However, in many patients with pustular psoriasis no underlying mutations are detectable. Furthermore, there is no association of DITRA with the occurrence of plaque psoriasis, and it is important to differentiate between DITRA and other types of pustular psoriasis, such as palmoplantar psoriasis. Histopathology revealed massive neutrophilic infiltration of epidermis and dermis (61). Immunohistochemistry showed IL-36γ in epidermal keratinocytes and absence of IL36RA in lesional skin of patients with DITRA (61, 66). Interestingly, biological inhibition of TNF-alpha, IL-12/23 and IL-17 seems to be more effective than anti-IL-1 treatment in patients with DITRA (67). Also, a monoclonal antibody against the IL-36 receptor (Spesolimab, BI 655130) has been shown to be efficacious in patients with generalized pustular psoriasis regardless of their mutation status (68).
Fig. 4. (A and B) Front and back of a female patient with acute flare of generalized pustular psoriasis presenting with erythroderma, pustules, pustular lakes and erosions.
Microbial molecules from viruses, bacteria or parasites are recognized by pattern recognition receptors and drive the expression of IFN via activation of downstream signalling (69). IFNs are a family of signal proteins that are released in an autocrine or paracrine manner by host cells to regulate and activate immune response. They are classified into 3 groups, type I IFN, type II IFN and type III IFN, and are differently produced (70). Type I IFN, represented by 13 subtypes of IFN-α and a single IFN-β, is ubiquitously produced, while type II IFN is produced by T cells and type III IFN by epithelial cells. In addition, type I IFN plays a crucial role in antiviral immunity and has been part of the standard treatment of hepatitis C and hepatitis B infections in recent years. Upon releasing, IFNs bind to different kinds of surface receptors, resulting in the activation of the JAK-STAT signalling pathway. Hence, activated STAT complexes act as intracellular transcription factors and regulate the expression of interferon-stimulated genes which are involved in cellular immunity, proliferation, differentiation and apoptosis (71).
Interferonopathies are a group of monogenic disorders defined by impaired interferon-mediated immune responses and upregulated interferon gene expression.
Stimulator of interferon genes-associated vasculopathy with onset in infancy (SAVI)
Stimulator of interferon genes (STING), encoded by the TMEM173 gene, is an endoplasmatic reticulum transmembrane protein that exists as a homodimer (72). It functions as an adapter that is essential for interferon-β (IFN-β) induction. Binding to its ligands, cyclic dinucleotides, triggers conformational changes leading to phosphorylation of TANK-binding kinase 1 and interferon regulatory factor 3 (IRF-3). Then, phosphorylated IRF-3 translocates into the nucleus and mediates the expression of IFNB1 (interferon-β) (Fig. 1) (73). Gain-of-function mutations within TMEM173 causes STING-associated vasculopathy with onset in infancy (SAVI) by constitutive STING activation, resulting in an increase and chronic hypersecretion of IFN-β (74). SAVI presents with recurrent fevers, interstitial lung disease, failure to thrive and systemic inflammation (Table I). However, the main clinical finding is vasculopathy (72).
Regarding the skin, patients with SAVI initially present with teleangiectatic, blistering and/or pustular rashes, mainly distributed on the fingers, toes, soles, cheeks and nose. Cutaneous symptoms start in the first weeks or months after birth, worsen by cold exposition, and can progress to severe ulcerative lesions due to peripheral vascular inflammation. Chronic involvement of the skin can manifest as acral violaceous plaques or nodules, and includes nail dystrophy, distal gangrenes and nasal septum perforations. These symptoms result from further vascular and tissue damage (72). Histological examination of lesional skin samples shows small vessel vasculitis. IgM, C3 and fibrin deposition was observed in lesional skin of single SAVI patients, indicative of an immune-complex-mediated mechanism (72). Given the pathogenic mechanisms in SAVI, inhibition of Janus kinase with blockade of type 1 IFN signalling is a promising treatment option. Both baricitinib and tofacitinib had favourable effects on skin and systemic symptoms in patients with SAVI (75, 76).
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature/proteasome-associated autoinflammatory syndrome
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE), also known as proteasome-associated autoinflammatory syndrome (PRAAS), is an autosomal recessive genetic disorder that affects the skin and subcutaneous tissue and presents with systemic inflammation. It is caused by mutations in proteasome or immunoproteasome subunit genes (PSMB3, PSMB4, PSMB8, PSMB9, POMP) (77–81).
CANDLE is not a primary interferonopathy, but is characterized by a proteasome – immunoproteasome dysfunction, leading to constitutional hypersecretion of type 1 IFNs (82). The proteasome and immunoproteasome are responsible for the degradation of impaired ubiquitinated cellular proteins by proteolysis (Fig. 1). In patients with CANDLE, proteasome and immunoproteasome dysfunction leads to an intracellular accumulation of ubiquitinated protein. The resulting cellular stress induces type I IFN genes to enhance IFN signalling and IFN synthesis. IFNs modulate the release and production of pro-inflammatory cytokines and cell recruitment, which culminates in further organ inflammation. Infections or cold exposure are potent trigger factors that can aggravate proteasome and immunoproteasome dysfunction.
Systemic symptoms in patients with CANDLE include growth delay, musculoskeletal symptoms and hepatosplenomegaly. Skin symptoms accompanied by fever are often the initial clinical manifestations of CANDLE, with onset in early infancy (Table I) (83). Mostly, they are located on the fingers, toes, ears and nose, and may be cold-triggered as reported for patients with SAVI. Initially, they present with periodic erythematous to purplish oedematous plaques that resemble perniotic lesions and can be accompanied by localized swelling (Fig. 5). The skin symptoms change over the course of the disease. With increasing age, transient annular, purpuric plaques with raised borders become the more common finding. In contrast to SAVI, tissue destruction with ulceration, perforation and development of gangrene is uncommon. Furthermore, a persisting violaceous periorbital and perioral oedema occurs (84, p. 438). Later on, progressive lipodystrophy is a main characteristic of patients with CANDLE. It starts in the face and progresses to involve the trunk and the extremities (77, 83).
Lesional skin biopsies revealed dense mixed infiltrates of mononuclear cells with irregular nuclei, atypical myeloid cells, but also some mature lymphocytes, neutrophils and eosinophils in the dermis. It has been postulated that the atypical myeloid cells are recruited by increased release of IFN from the bone marrow, and that they further infiltrate peripheral organs (85). Immunohistochemistry demonstrated dermal myeloperoxidase- and CD68-positive myeloid cell infiltrates in patients with CANDLE (77, 85, 86). In addition, CD163-positive histiocytes, as well as CD123-positive plasmacytoid dendritic cells, were observed (85).
In line with SAVI, patients with CANDLE benefit from inhibition of Janus kinase. This underlines the pathophysiological role of type 1 IFN signalling (75).
Fig. 5. Skin symptoms in an infant with chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature. (A) Erythematous papules and plaques on the right foot and right lower leg. (B) Purplish oedematous plaques on the fingers that resemble perniotic lesions with accompanying swelling of the hand.
The NOD2 pathway is involved in the innate immune defence against invading pathogens. NOD2 is a member of a family of pattern recognition molecules and is mainly expressed by antigen-presenting cells and intestinal Paneth cells (87, 88). It contains 2 N-terminal CARD domains for downstream signalling through CARD-CARD interaction, a NOD/NACHT domain with ATPase activity and a C-terminal domain comprised of 10 LRR motifs (89). In addition, the LRR domain provides a binding-site for its natural ligand muramyl dipeptide (MDP), a degradation product of ubiquitous peptidoglycan (90). Without a stimulus, NOD2 is silenced via auto-inhibition. The engagement of NOD2 and MDP induces a conformational change and oligomerizes the exposed NOD/NACHT domain. This leads to NOD2 activation and recruitment of the serine/threonine kinase receptor-interacting protein kinase 2 (RIP2) (91). The CARD-CARD interaction between NOD2 and RIP2 promotes the activation of NF-κB and mitogen-activated protein kinase, resulting in production of inflammatory cytokines, chemokines and adhesion molecules (Fig. 1) (92).
Blau syndrome
Blau syndrome is a NOD2-associated granulomatous inflammatory disease with an autosomal dominant inheritance that usually starts between infancy and the age of 5 years (93). Several NOD2 gain-of-function mutations were described to cause Blau syndrome, most of them were reported in the NOD/NACHT domain (93–96). The main clinical characteristics are arthritis, skin inflammation and uveitis (Table I; Fig. 6) (97).
In infancy, patients with Blau syndrome show a monomorphic micropapular erythematous rash with fine desquamation as the initial symptom (84, p. 373–374). The rash often starts on the dorsal trunk and further affects face and extremities. Over time, the initially erythematous rash becomes brownish and scaly. Furthermore, patients can develop subcutaneous nodules on the lower extremities, mimicking erythema nodosum (98). In single cases, skin affection, such as erysipelas-like lesions, urticarial rash, livedoid lesions and vasculitis, have been observed (99–101). Histologically, the skin lesions are characterized by naked sarcoidal granuloma formation (100).
Fig. 6. Blau syndrome. A) A 12-year-old boy with disseminated small scaly solid papules with onset at age 6 months. These asymptomatic eruptions improve spontaneously, but relapse again without specific events. (B) A 4-year-old boy showing joint involvement with cystic swelling of the dorsal sides of the left hand.
Nucleotide oligomerization domain (NOD)-like receptor family CARD domain-containing protein 4 (NLRC4) and NLCR4 inflammasomopathies
The NLRC4 inflammasome is activated by at least 2 compounds of Gram-negative bacteria, flagellin and type 3 secretion protein (T3SS) (102–104). However, the interaction between ligand and NLRC4 does not occur directly. Instead, the sensor protein NLR family of apoptosis inhibitory protein (NAIP) physically binds flagellin or T3SS and co-assembles with NLRC4, leading to its activation through conformational change (105). Furthermore, studies have shown that phosphorylation by the kinase Pkcδ is required for the complete NLCR4 activation (106). Once activated, NAIP-NLRC4 forms a multimeric complex, known as inflammasome, which recruits and activates caspase-1 (CASP-1) (107). CASP-1 is involved in anti-bacterial responses by triggering pyroptosis, a form of inflammatory cell death (108). In addition, it mediates the processing and release of IL-1β and IL-18 (Fig. 1) (109, 110).
Gain-of-function mutations within the NLCR4 gene are linked to NLCR4 inflammasomopathies (111, 112). These autosomal dominantly-inherited mutations promote the spontaneous formation of the NLCR4, inflammatory cell death and production of IL-1β and IL-18 (113). The clinical spectrum is manifested by a variety of symptoms, and can range between mild CAPS-like phenotypes with urticarial rash and little inflammation as well as severe conditions of macrophage activation syndrome and enterocolitis with onset in infancy (Table I) (111, 112). Macrophage activation syndrome comprises a life-threatening condition of fever, hyperferritinaemia, hepatobiliary dysfunction and haemophagocytosis. Disseminated intravascular coagulation and acute respiratory distress syndrome may occur (111).
With respect to the skin, patients present with erythematous or urticarial rashes. In contrast to patients with CAPS, the lesional skin of NLRC4 inflammasomopathies patients is characterized by a lymphohistiocytic infiltrate (114). As serum IL-18 levels are markedly increased in NLRC4 inflammasomopathies compared with patients with CAPS, it could be speculated that IL-18 may mediate cutaneous recruitment of lymphocytes and macrophages (112).
PLCG2-associated antibody deficiency and immune dysregulation and autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation
Phospholipase C-gamma 2 (PLCγ2)-associated antibody deficiency and immune dysregulation (PLAID) and autoinflammation and PLCγ2-associated antibody deficiency and immune dysregulation (APLAID) are autosomal dominant syndromes, which are based on mutations in PLCG2 (115, 116). In-frame deletions of exon 19 and exons 20–22 are known for PLAID (115). APLAID is induced by the S707Y mutation in PLCG2 (116).
PLCγ2 is a transmembrane phospholipase enzyme that catalyses the hydrolysis of phosphatidylinositol bisphosphate (PIP2) to diacylglycerol (DAG) and inositoltriphosphate (IP3). IP3 modulates the calcium release as a second messenger from the endoplasmic reticulum and thereby mediates cellular signal transduction. PLCγ2 regulates various cellular functions, such as protein transport, apoptosis/cell survival, migration and immune responses (Fig. 1). It is expressed mainly in lymphoid and myeloid cells. In patients with PLAID, PLCG2 deletions alter the carboxyl-terminal Src homology 2 domain (SH2), which is critical for PLCγ2-autoinhibtion. As a result, PLCγ2 is constitutively activated, but with reduced intracellular signalling at physiological temperatures (115). In APLAID, the S707Y substitution within the autoinhibitory SH2 domain leads to hyperactivation of the PLCγ2 enzyme and exhibits exactly the opposite effects, with increased cellular signalling at physiological temperatures (115, 116). Interestingly, it has been shown that this PLCγ2 hyperactivation results in enhanced NLRP3 inflammasome activity via intracellular Ca2+ signalling (117).
PLAID and APLAID comprise a wide spectrum of clinical and laboratory findings. Both present with an increased susceptibility to recurrent infections and variable atopic features and/or autoimmune phenomena.
Regarding the skin, patients with PLAID present with pruritic wheals or erythematous patches since birth. Skin lesions are provoked by cold air or evaporative cooling of the skin and last from minutes to hours (115). In some cases, oesophageal burning sensations after consumption of cold/frozen foods were reported (115, 118). Furthermore, syncopal episodes were described, when completely exposed to cold water (118). The urticarial lesions are based on cold-induced mast cell activation with consecutive degranulation (115). In addition, PLAID can present with neonatal-onset acral ulcerative granulomatous skin lesions (fingers, nose and ears), which may or may not disappear during childhood. Ulcers are often haemorrhagic and can affect the subjacent tissue, such as erosion of the nasal cartilage. The granulomas are characterized by CD68+ histiocytic infiltrates with multinucleated giant cells, a mild CD4/CD8 lymphocytic infiltrate and scattered eosinophils (118).
In contrast to PLAID, patients with APLAID do not show any cold-induced symptoms, but present with epidermolysis bullosa-like eruptions in early childhood. Over time, patients with APLAID develop recurrent erythematous plaques and vesiculopustular skin lesions, which worsen after heat, sun exposure and pressure (116). The underlying pathophysiological mechanisms remain to be elucidated.
All autoinflammatory skin diseases have in common that they occur in flares of systemic inflammation with elevated acute phase reactants and characteristic clinical symptoms. Many are caused by gene mutations that impact critical immune responses resulting in autoinflammation, but also autoimmunity and immunodeficiency. The identification of various genetic variants has broadened our understanding of host defence mechanisms and their interactions. Still, the recognition of dermatological phenotypes and clinical presentation of patients with autoinflammatory diseases are crucial for diagnosis and treatment. The occurrence of skin lesions as the only symptom is exceptional and associated complaints and symptoms should always be assessed. In most conditions, inflammatory markers, such as CRP, ESR, SAA and S100 proteins, are elevated. Although these are non-specific findings, they may prompt further investigations towards autoinflammatory disorders, such as genetic analyses in early-onset and/or familial cases.
Targeted inhibition of cytokines is effective in many of these disorders and has significantly improved the health-related quality of life of patients. Based on a better pathomechanistic understanding, novel small molecules (e.g. inflammasome inhibitors) are currently being developed (119) and may enable even more precise therapies. In addition, novel technologies such as CRISPR/Cas may enable targeted gene therapy in autoinflammatory diseases.


 Click to show fullsize
Click to show fullsize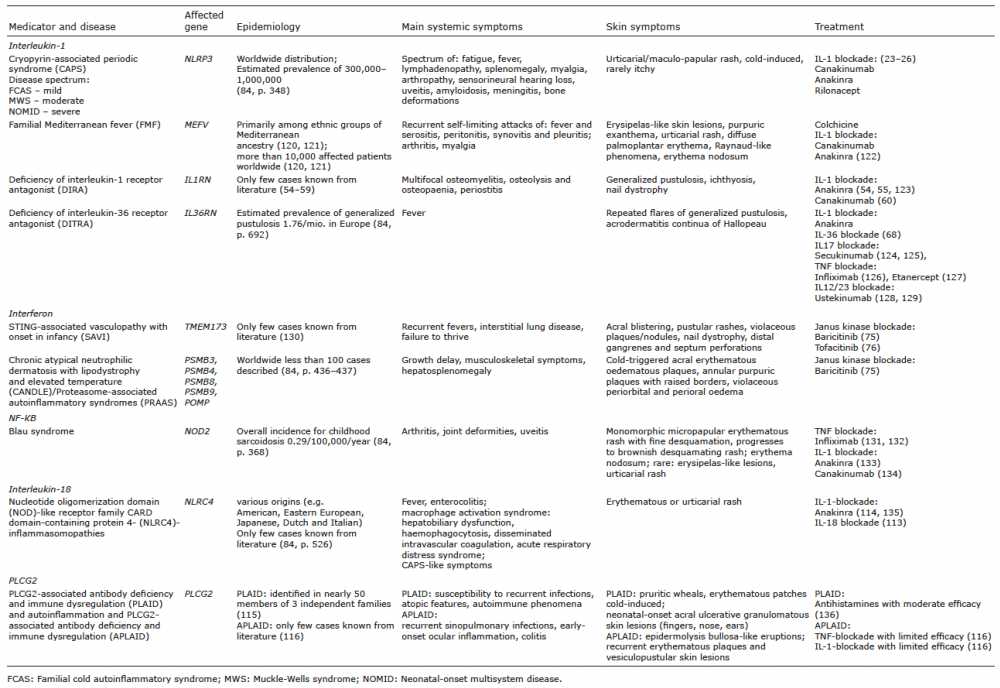 Click to show fullsize
Click to show fullsize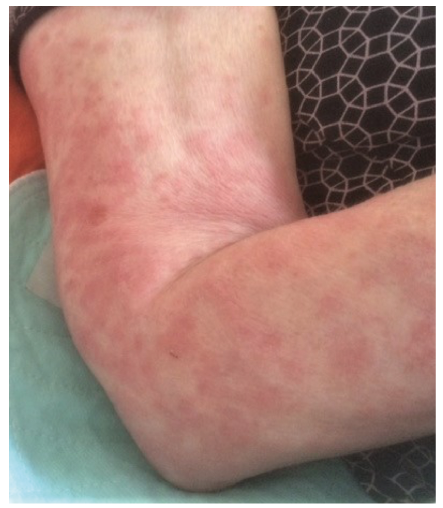 Click to show fullsize
Click to show fullsize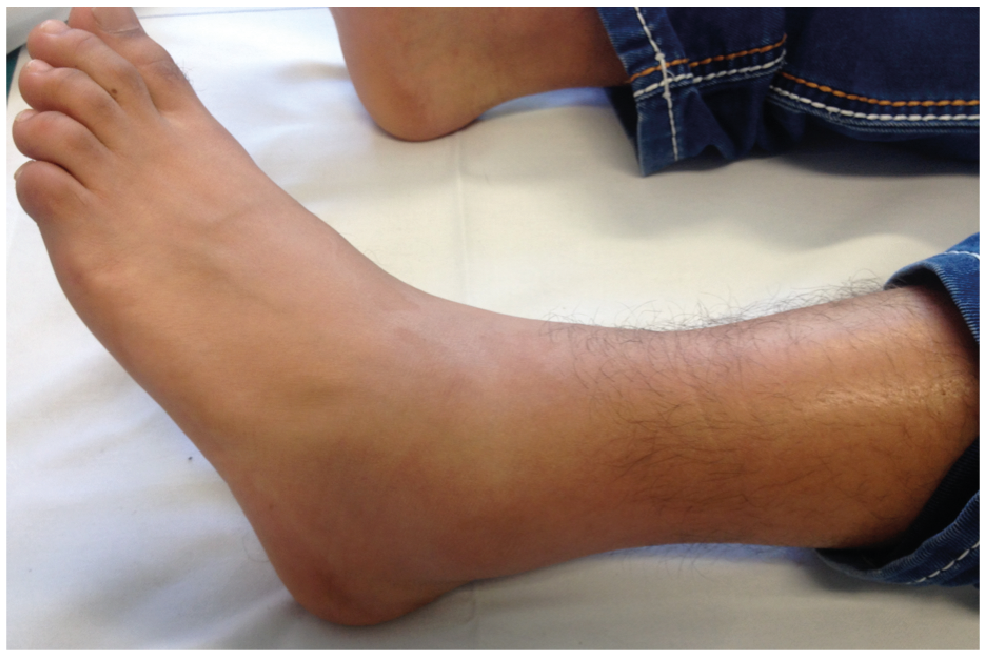 Click to show fullsize
Click to show fullsize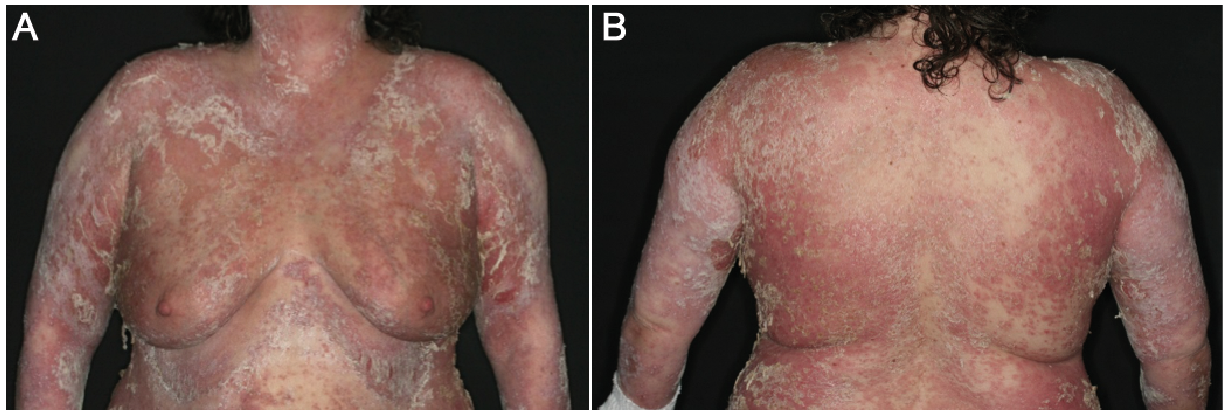 Click to show fullsize
Click to show fullsize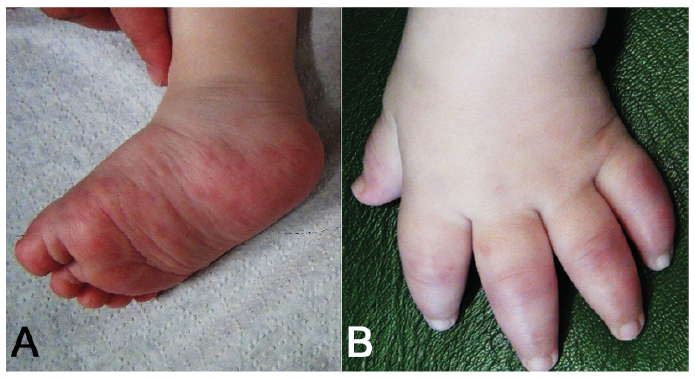 Click to show fullsize
Click to show fullsize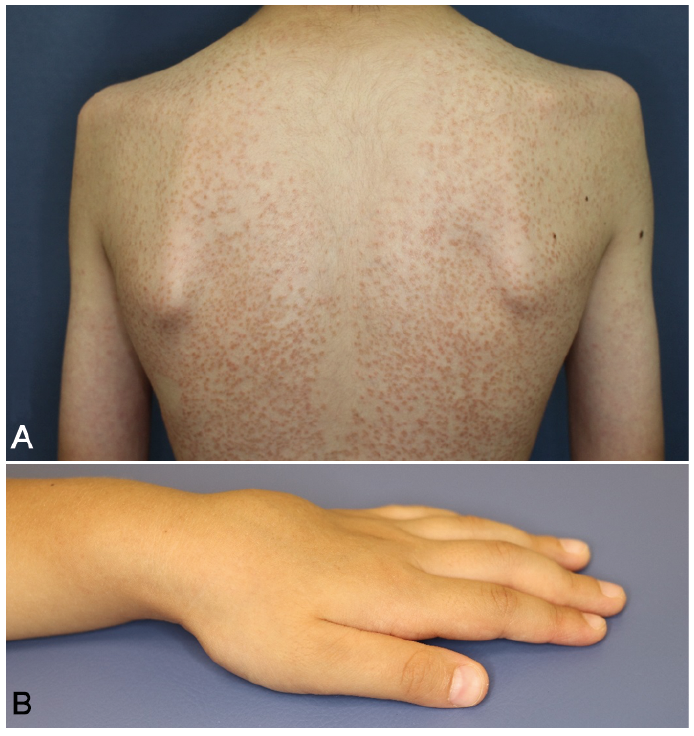 Click to show fullsize
Click to show fullsize