


1Centre for Cell Biology and Cutaneous Research, The Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, and 2Department of Dermatology, Royal London Hospital, Barts Health NHS Trust, ERN-Skin, London, UK
Inherited monogenic palmoplantar keratodermas are a heterogeneous group of conditions characterised by persistent epidermal thickening of the palmoplantar skin. Palmoplantar keratodermas are grouped depending on the morphology of the keratoderma into diffuse, focal/striate or papular/punctate. Some palmoplantar keratodermas just affect the skin of the palms and soles and others have associated syndromic features which include changes in hair, teeth, nails, hearing loss or cardiomyopathy. Next generation sequencing has helped discover genes involved in many of these conditions and has led to reclassification of some palmoplantar keratodermas. In this review, we discuss the diagnostic features of palmoplantar keratodermas and management options.
Key words: keratoderma; palmoplantar; keratin; genetic; inherited.
Accepted Feb 12, 2020; Epub ahead of print Mar 9, 2020
Acta Derm Venereol 2020; 100: adv00094.
Corr: Prof. Edel A. O’Toole, Centre for Cell Biology and Cutaneous Research, The Blizard Institute, Barts and the London School of Medicine and Dentistry, 4 Newark Str, London E1 2AT, UK. E-mail: e.a.otoole@qmul.ac.uk
The palmoplantar keratodermas are a complex group of diseases where the main feature is thickening of the skin of the palms and soles. Genetic testing has given insight into the biology of these conditions and has allowed experts to reclassify them. In this review, we present a summary of the key features of the major types of palmoplantar keratodermas and discuss their management.
The palmoplantar keratodermas (PPK) are a complex group of conditions that are characterised by persistent epidermal thickening (hyperkeratosis) of palmoplantar skin. The PPK are traditionally classified as hereditary (HPPK) or acquired. The main feature distinguishing hereditary from acquired PPK is the presence of a positive family history, early onset of disease, associated syndromic features and relative treatment resistance (1). Sporadic (spontaneous) mutations need to be considered in those without a family history or late onset disease (2).
Next generation sequencing has given us a better understanding of HPPK pathophysiology and has shown that one genotype can have several phenotypes. This has led to reclassification of some PPK thought previously to be distinct entities. Laboratory investigation shows that palmoplantar skin is a site at which multiple molecular pathways converge: gap junctions via connexins, intracellular adhesion through desmosomes and mechanical stability by means of the keratin cytoskeleton amongst others (3).
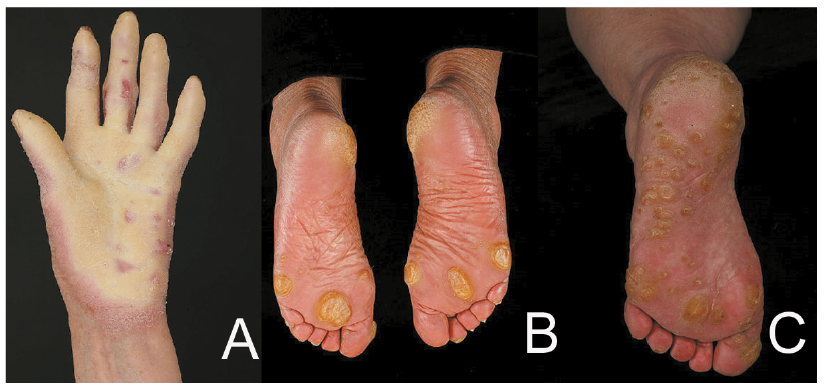
An initial approach to PPK is to take a history asking about age of onset, palmoplantar pain and/or blistering, sweating and infection and other associated features including hearing loss, abnormal hair, nail or teeth/ mucosal problems, cysts and family history including family history of cancer. Clinical examination can usually differentiate PPK into 3 groups: diffuse, focal or punctate (Fig. 1). The clinical features and management will be discussed in this review and are summarized in Table I.
Fig. 1. Patterns of palmoplantar keratodermas. A) diffuse, B) focal and C) punctate.
Table I. Summary of hereditary palmoplantar keratodermas (HPPK)
Diffuse epidermolytic PPK (EPPK; MIM# 144200, KRT9, KRT1) is the most common diffuse PPK with epidermolytic changes in suprabasal keratinocytes seen on histology (4). It is inherited in an autosomal dominant (AD) fashion due to mutations in KRT9 and sometimes KRT1 (5, 6). The KRT9 gene encodes for the type I keratin, keratin 9, which is mainly expressed in suprabasal palmoplantar skin. Type I keratins form heterodimers with type II keratins, in this instance, possibly keratin 1, found in the epidermis including the palms and soles, to form intermediate filaments which provide strength to the skin (7, 8).
This PPK develops in infancy and in adults the hyperkeratosis is brown-yellow and confluent with fissuring, confined to the palmoplantar surfaces with an erythematous edge. Limited transgradient lesions or flexural hyperkeratosis may indicate KRT1 mutations (9). There may be a history of blistering and knuckle pads have been reported.
Treatment is mainly by mechanical debridement and use of keratolytics like urea, salicylic acid and lactic acid in emollient, sometimes under occlusion. Oral retinoids can help but pain from increased fragility limits their use (10, 11). Topical calcipotriol has been reported to be of benefit (12). Small inhibitory RNA therapy may be a possibility for the future (13).
Non-epidermolytic PPK type Bothnia (MIM# 600231, AQP5) was first described in Northern Sweden and is due to heterozygous missense mutations in AQP5 (14). This gene encodes for the water-channel protein aquaporin-5, which is expressed in exocrine glands but also the plasma membrane of palmar stratum granulosum. Mutations in the gene allow these cells to transport water by forming open water channels at this site.
This PPK usually starts in the first few months of life and is classically a brown-yellow, smooth keratoderma with an erythematous edge. Due to the defect in aquaporins, water immersion leads to a white spongy appearance which lasts for about 30 min. Pitted keratolysis and dermatophyte superinfection is common and can be treated with topical erythromycin or oral anti-fungals (15). Acitretin at low doses can be helpful.
NEPPK type Nagashima (MIM# 615598, SERPINB7) is an autosomal recessive (AR) PPK due to mutations in SERPINB7 described in Japanese and Chinese patients. Mutations in this gene may cause uncontrolled activity of proteases in the stratum corneum leading to increased water permeation (16, 17).
The condition presents in early life and is characterised by mild hyperkeratosis and striking redness extending to the dorsum fingers/feet and anterior wrist (18). A white spongy appearance after water immersion is seen (19) and associated hyperhidrosis and bacterial/fungal superinfection can be present.
Mal de Meleda (MDM; MIM#248300, ARS) is an eponymous AR PPK named after the Island of Mljet (née Meleda) (20). Mutations in ARS which encodes SLURP-1 cause MDM (21). SLURP-1 stimulates nicotinic acetylcholine receptors which regulate keratinocyte growth. When SLURP-1 is not functioning, it is thought that there is a reduction in keratinocyte apoptosis regulation (22).
MDM is characterised by a diffuse, ivory-yellow macerated hyperkeratosis with a characteristic malodour and striking erythematous trangradiens that extends to dorsal surfaces. A key feature includes lesions on the elbows and knees (21). Perioral hyperkeratosis and erythema can be present (23). The disease starts in infancy and progresses through life. Flexion contractures can occur and constrictive bands can lead to spontaneous amputation (24). Nail thickening, subungual hyperkeratosis and koilonychia can be present. The diffuse keratoderma of Gamborg-Nielsen also due to ARS mutations is likely a mild variant of MDM (25). Interestingly female heterozygotes can also present with a mild phenotype (26).
Treatment of bacterial/fungal superinfection and the hyperhidrosis is helpful, although the mainstay of treatment is oral retinoids which improve the hyperkeratosis although the erythema may worsen (27, 28).
Loricrin keratoderma (LK; MIM# 604117, LOR) is AD and starts in early childhood. It is due to a mutation in LOR which interferes with the regulation of epidermal cornification (29). Some children are born with a collodion membrane and generalised scaling from birth may be noted (30). During childhood the PPK develops with a characteristic diffuse, honeycomb pattern which can extend to wrist/ankles and is associated with non-migratory red plaques on the extensor surfaces of joints (31). Trangradiens is present but the edges of the hyperkeratosis are ill-defined. Constrictive bands can develop. Knuckle pads may be present (32) and hearing is intact.
Isotretinoin has been reported as helpful (33). In the future, there may be a role for treating LK with vascular endothelial growth factor 2 receptor inhibitors (34).
Keratosis linearis with ichthyosis congenita and sclerosing keratoderma (KLICK, MIM#601952, POMP) presents in early childhood and similar to LK, starts with generalised erythema and fine scaling with the subsequent diffuse, smooth PPK (35). Inheritance of this PPK is AR caused by mutations in the POMP gene, which lead to endoplasmic reticular stress and subsequent dysfunctional profilaggrin processing (36, 37). Flexural linear and starfish keratoses overlying large joints are distinctive (35). Acitretin can be helpful for both the ichthyosis and keratoderma (38).
PPK with scleroatrophy (Huriez syndrome, MIM #181600) is a cancer-related PPK caused by haploinsufficiency in SMARCAD1 (39). Scleroatrophy is seen across the entire palm and fingers (40) with mild hyperkeratosis of the palms. The affected skin is often red and the palms are usually more severely affected than the soles. Hypoplastic nail changes may be present and 50% experience hypohidrosis. The most important characteristic is the 100-fold increased risk of developing squamous cell carcinoma (SCC) in the affected skin. Acitretin may be helpful for the PPK and prevention of SCC (41).
Palmoplantar hyperkeratosis with squamous cell carcinoma of skin and sex reversal (MIM#610644, RSPO1) is similar to Huriez syndrome as it is a mild PPK with sclerodactyly and nail hypoplasia (42). This condition is AR caused by mutations in RSPO1 (43). This gene is responsible for stabilising β-catenin in the Wnt signaling pathway which antagonises SRY/SOX9 actions for sex determination (44). A characteristic feature is the female to male sex reversal seen in females. The karyotype is 46, XX. Predisposition for cutaneous SCC and also laryngeal SCC is noted (45). Periodontitis with loss of teeth may be present.
Odonto-onycho-dermal dysplasia spectrum (OODD) is an AR condition caused by mutations in WNT10A which starts in early life (46). In absence of WNT10A, β-catenin pathway activity and epithelial progenitor proliferation are reduced. In these patients, Wnt-active stem cells are seen in sweat ducts, hair follicles, nails and taste buds and there are differentiation abnormalities in palmoplantar skin (47).
Typically the PPK is mild, diffuse and erythematous with late onset palmoplantar hyperhidrosis (48). There is overlap with Schöpf-Schulz-Passarge syndrome (SSPS) and patients can have hypodontia with abnormal teeth, nail hypoplasia, smooth tongue and hypotrichosis (49). Eyelid hidrocystomas and other benign adnexal tumours can present at a later age (50). Biopsy of palmoplantar skin shows eccrine syringoadenomatosis (46). Tumours/cysts may need treatment with surgery or laser.
Olmsted syndrome (OLS; MIM# 614594 - TRPV3, 300918 - MTBSP2) typically presents as a severe mutilating transgradient keratoderma. AD, AR, semi-dominant and X-linked recessive (XLR) forms have been described caused by mutations in TRPV3 (AD, AR) and MBTPS2 (XLR) (51, 52) The Ca2+-permeable cation channel TRPV3 is expressed abundantly in keratinocytes, associated with TGF-α/EGFR signalling and may play a role in keratinocyte differentiation by elevating Ca2+ within these cells (53). MBTPS2 mutations in skin may cause a decrease in responsiveness to sterols subsequent to depletion of proteases (54).
The keratoderma is diffuse and can be associated with digital flexion deformities and constrictive bands. Periorificial/ear/nose/umbilical keratoses can also be present. Dystrophy of teeth, nails and cornea, alopecia, erythromelalgia and joint laxity have also been reported. A milder phenotype can simulate pachyonychia congenita (PC) (55). Melanoma and SCC have been reported in OLS (56).
Treatment in general is difficult with variable response to systemic retinoids. Topical anti-inflammatories can be helpful for hyperkeratosis and itching. Surgery with excision and grafting of the keratoderma can lead to more favourable long-term outcomes (2). Finally, there has been one report of a patient treated with the EGFR inhibitor, erlotinib, which gave a transient improvement (57).
PPK with periodontitis (MIM#245000, allelic disease: Haim-Munk #245010, CTSC) encapsulates both Papillon-Lefèvre syndrome (PLS) and Haim-Munk syndrome (HMS). Both conditions are caused by homozygous mutations in CTSC. CTSC is expressed in the palms, soles, alveolar bone and keratinized gingiva; it plays a role in immune cell protease activation and possibly has a role in epidermal differentiation leading to this particular phenotype (58, 59).
Patients have thickening and erythema of the palmoplantar skin, associated with bacterial skin infections and periodontitis (60). The PPK typically starts/worsens with the breakthrough of the deciduous teeth and actually improves after tooth loss/reduction of gingival inflammation (61). Hyperkeratotic plaques on the extensor surfaces are seen. PLS is associated with pyogenic liver abscesses (62). HMS has the same features with arachnodactyly, onychogryphosis and acro-osteolysis, mainly described in Cochin Jews (63).
Retinoids have shown to improve the PPK and oral disease (62). Specialist dental care is essential. For those above the age of 12, low dose tetracycline may be helpful for gingivitis, even at subtherapeutic doses (64).
Cerebral dysgenesis, neuropathy, ichthyosis and PPK syndrome (CEDNIK; MIM#609528, SNAP29) is a PPK with neurological manifestations which starts in infancy. This AR condition is caused by mutations in SNAP29 which lead to abnormal lamellar granule formation with subsequent aberrant epidermal differentiation (65). Around one year of age a diffuse keratoderma and ichthyosis become apparent (65, 66). Histology of CEDNIK demonstrates clear vesicles in the top 3 layers of the epidermis. Treatment is symptomatic.
Autosomal recessive keratoderma ichthyosis and deafness (ARKID) is caused by mutations in VPS33B. Mutations in this gene can lead to abnormal lamellar body morphology and function and impaired barrier formation. Patients present with progressive hearing loss (normal at birth) and delayed development. The PPK that develops is diffuse and associated with flexion deformities and autoamputation (67).
PPK, leukonychia and exuberant scalp hair is caused by AR mutations in FAM83G. FAM83G may have a role as a suppressor of the Wnt signalling pathway. Diffuse, verrucous thickening of soles and mild palmar involvement is noted. Leukonychia/dystrophy of the toenails and rapid hair growth are also seen (68).
Striate PPK (PPKS) can be separated into PPKS1 (MIM# 148700, DSG1) (69), PPKS2 (MIM# 612908, DSP1) (70), and PPKS3 (MIM# 607654, KRT1) (71). The DSG1 (desmoglein 1) and DSP1 (desmoplakin) genes encode for desmosomal proteins required for intercellular adhesion of keratinocytes (72). Mutations in the V2 domain of KRT1 cause PPKS3 and disrupt the intermediate filament network.
Classically, striate PPK presents with linear bands of hyperkeratosis on the palmar surface (73). Diffuse or focal changes may also be present. It is usual for plantar changes to be focal and to present early in life (i.e. first or second year) and the palmar changes follow (71). If patients exhibit woolly/curly hair or abnormal dentition associated cardiomyopathy should be considered. Histology in PPKS can be helpful as it will demonstrate acantholysis of keratinocytes pointing to a desmosomal mutation (74).
Tylosis with oesophageal cancer (TOC; MIM#148500, RHBDF2) is rare condition that is AD and caused by gain of function mutations in RHBDF2 which create a hyperproliferative phenotype through continuous EGFR signalling (75). Patients present with focal keratoderma at sites of pressure usually by 8 years of age. Patients also have follicular hyperkeratosis and oral leukokeratosis similar to PC (76). Most patients with tylosis have a family history of oesophageal carcinoma and carry a risk of oesophageal cancer of 95% by age 65 years (77). Regular screening for oesophageal dysplasia is required and smoking and alcohol should be avoided.
Tyrosinaemia type II (MIM#276600, TAT) (78) is a very rare AR condition that initially presents in the first few months of life with ocular symptoms including photophobia and pain and subsequent ocular scarring (79). Hyperkeratosis of the palms that follows the fingerprints develops prior to a focal plantar keratoderma (80). About 50% of patients will have some form of intellectual disability and neurological signs. Increased tyrosine levels found in the bloods/urine, due to abnormal tyrosine aminotransferase function, can aid diagnosis (79) and symptoms may be prevented by a phenylalanine/tyrosine free diet.
Pachyonychia congenita (PC; Multiple MIM#) is a heterogeneous groups of conditions characterised by nail dystrophy and a painful focal keratoderma. The Pachyonychia Project (www.pachyonychia.org) has collected extensive data on PC collated in the International PC Research Registry (IPCRR). The current classification is based on keratin gene mutation: PC-6a, PC-6b, PC-6c, PC-16 and PC-17 (81). These 5 subtypes have replaced the original PC type 1 and 2 classification. Mutations in these genes lead to increased palmoplantar skin fragility due to disruption of keratin filament formation, nail changes and changes in the pilosebaceous unit.
The IPCRR data has shown that 90% of patients > 3 years old will have 3 clinical features: toenail dystrophy, plantar keratoderma and plantar pain (82). The hypertrophic nail dystrophy starts in the first few months of life up to 9 years. KRT6A mutations are associated with early onset disease. All nails need not be affected. The focal plantar keratoderma starts when children begin to weight bear with blistering under the calluses (83). Plantar pain has a neuropathic component and can be severe enough to require ambulatory aids. The palmar lesions are usually less prominent than the plantar lesions, except in the case of PC-KRT16 with striate lesions (82).
Follicular hyperkeratoses are seen in areas of friction. Oral leukokeratosis can resemble oral candidiasis and laryngeal involvement can lead to hoarseness and infantile respiratory obstruction (83). Cysts occur in all subtypes of PC although KRT17 mutations are usually associated with more steatocystomas/pilosebaceous cysts and natal teeth (less commonly KRT6A) (83). KRT6A mutations can also be associated with ear pain with feeding difficulties in infants/toddlers. PC-6C has a limited keratoderma and mild nail dystrophy (84).
Current treatment is largely mechanical paring of the calluses, assisted by a podiatrist, if necessary. Low dose acitretin can help in some patients but is associated with increased pain. Botox injections can also help reduce pain (85). The IPCC has had some promising results with siRNA and rapamycin (86, 87). Clinical trials of topical rapamycin are ongoing. As with most PPKs, comfortable foot wear and customised insoles are helpful.
Hypotrichosis-osteolysis-periodontitis-palmoplantar keratoderma syndrome (HOPP; MIM 607658) is a rare syndrome with a phenotype similar to PLS/HMS although CTSC mutations were not found. There is a striking keratoderma with a reticular pitted/punctate pattern (88). Progressive hypotrichosis from 6 years of age is seen sometimes with pili annulati. Lingua plicata can be noted at an early age (89).
PPK-deafness syndromes are mainly caused by GJB2 and rarely GJB6 mutations. Numerous gap junctions are present in the skin and inner ear. Mutations in gap junction genes lead to abnormal keratinocyte differentiation/growth and dyfunctional inner ear potassium ion recycling required for hearing (90).
Phenotypically these PPK-deafness syndrome are distinct and still carry their eponymous names. Despite having mutations in the same gene, keratitis-ichthyosis-deafness-like (MIM# 148210), hystrix-like ichthyosis-deafness (MIM#602540), palmoplantar keratoderma-deafness (MIM#148350), Bart-Pumphrey (MIM# 149200) and Vohwinkel (MIM#124500) syndromes have phenotypic differences likely explained by mutations in particular domains of connexin 26 (GJB2) (91). Cardinal features are PPK and hearing loss of varying severity. For example, Vohwinkel syndrome has marginal translucent papules which become confluent over time. It also has the ‘classic’ starfish keratoses on the knuckles and extensor surfaces of joints and pseudoainhum (92).
Oral retinoids are helpful for the constricting bands seen in Vohwinkel syndrome (93) but surgery may be required.
PPK and cardiomyopathy are similar to PPKS1&2 as they are also associated with keratinocyte disadhesion. Naxos syndrome (AR) caused by mutations in JUP (MIM# 601214) encoding plakoglobin presents with woolly hair at birth followed by a diffuse/striate keratoderma in the first year of life. Cardiomyopathy presents in adolescence and has 100% penetrance (94). Carvajal-Heurta syndrome (CHS), caused by mutations in DSP (MIM# 605676), is like Naxos although the cardiomyopathy presents earlier in the teens and is usually biventricular. Some patients with CHS have short woolly hair and keratoses on the elbows/knees (95, 96). The DSP and JUP genes encode desmosomal proteins required for formation of cell junctions in hair, skin and cardiac tissue (97). Mutations in KANK2 can cause woolly hair, hypotrichosis and a PPK without cardiac involvement (98). The KANK2 gene regulates steroid receptor coactivators. Patients with a striate keratoderma/PPK and woolly hair should have cardiac investigations. Family members should also be screened as these can have AR or AD inheritance.
Punctate PPK occurs in 1 in 100,000 people and has AD inheritance. Mutations in the AAGAB gene occur in about 1/3 (99). The AAGAB gene is involved in recycling of EGFR proteins and impairment in this function leads to keratinocyte proliferation (100). Also, mutations in COL14A1, encoding collagen XIV required for fibrillogenesis, have been found in Chinese families (101). Lesions seem to develop after the teenage years. Lesions are typically papular sometimes coalescing into plaques (102). The lesions are worse in manual labourers. Rarely, there is an association with malignancy (99). Treatment with mechanical debridement is helpful. Comfortable shoes are key. Acitretin and alitretinoin can be helpful for some (103).
Marginal papular keratoderma describes acrokeratoelastoidosis (AKE) and focal acral hyperkeratosis (FAH), thought to be inherited in an AD manner. AKE is characterized by small crateriform papules along ‘Wallace’s’ line on the medial aspect of the foot and the border of the palmar thenar/hypothenar eminences (104). FAH, differentiated by the lack of fragmented dermal elastic fibres on histology, is associated with knuckle pads and hyperkeratosis extending onto the Achilles tendon (105) presenting in the teenage years in individuals of African or Afro-Caribbean ethnicity.
Transient Aquagenic keratoderma (TAK) is an unusual keratoderma that mainly effects the palms and is triggered by contact with water or sweat. Patients are typically young women and after a few minutes of exposure to water a fine white papular eruption is present on the palms (106). The eruption resolves after drying, leaving minimal hyperkeratosis. TAK can be differentiated from hereditary papulotranslucent acrokeratoderma (MIM 101840) as the papules in TAK do not persist. Aquagenic wrinkling of the palms, seen in 50% of patients with cystic fibrosis and 10% of CFTR mutation heterozygotes, can also look similar (107).
Cole disease (MIM# 615522, ENPP1) is a very rare genodermatosis characterised by congenital or early-onset punctate keratoderma (108). The condition can be AD and AR and is due to ENPP1 mutations which impair homodimerization of the ENPP1 protein leading to impaired melanocyte regulation and function (109).
Over time children develop well-defined hypopigmented macules which are most prominent on the extremities. Cases of associated calcinosis cutis or tendon calcification have been reported.
PLACK syndrome (MIM# 616295, CAST) is an AR disorder characterised by peeling skin, acral keratoses, leukonychia, cheilitis and knuckle pads causes by mutations in CAST which causes dysregulation of keratinocyte adhesion and apoptosis (110).
The PPK are a heterogeneous group of conditions with a biologically fascinating diversity of genetic mutations. Modern sequencing techniques have aided our ability to re-classify these conditions. Targeted gene sequencing and keratoderma specific gene panels will aid in confirming diagnoses.
EAO is on the MSAB of Pachyonychia Project.


 Click to show fullsize
Click to show fullsize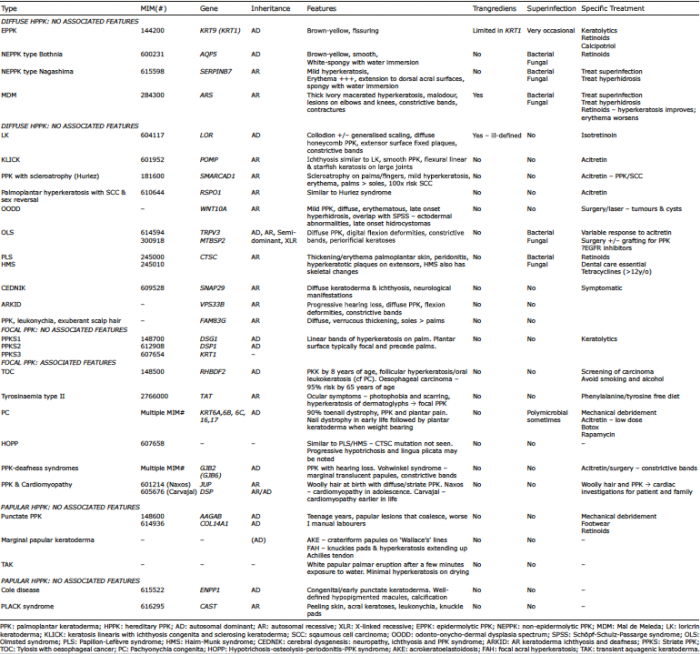 Click to show fullsize
Click to show fullsize