


Department of Dermatology, Ruijin Hospital, School of Medicine, Jiao Tong University, Shanghai 200025, China. E-mail: jie-zheng2001@126.com
Accepted Jun 4, 2020; Epub ahead of print Jun 9, 2020
Acta Derm Venereol 2020; 100: adv00192
Chronic active Epstein-Barr virus infection (CAEBV) is one of the EBV-associated T- and NK-cell lymphoproliferative disorders. Hydroa vacciniforme-like lymphoproliferative disorder (HVLLPD) is a sub-type of CAEBV, characterized by facial oedema and recurrent necrotic vesiculopapules in sun-exposed area, hypersensitivity to mosquito bites and extracutaneous manifestations, such as fever, fatigue, lymphadenopathy, hepatomegaly, etc.
To date, different therapeutic approaches, such as haematopoietic stem cell transplantation (HSCT), chemotherapy, immunosuppressor and immunomodulator, have been tested to treat HVLLPD, as there is a lack of a standard approach. In patients with HVLLPD, it can be difficult to distinguish between benign lymphoproliferative disease and lymphoma, which is observed in other EBV-associated T/natural killer cell lymphoproliferative disorders (1). Thus, it is challenging to determine whether HSCT or chemotherapy is suitable for these patients. Meanwhile, chemotherapy and preparative regimens of HSCT may lead to severe complications, even death, and sometimes EBV lytic reactivation. However, each episode is either sudden or severe. A treatment that can control the inflammation of each episode and slow down progression from benign lymphoproliferation to lymphoma needs to be found. We chose intravenous immunoglobulin (IVIG), which is a safe and ideal choice, as it has anti-viral (2) and anti-inflammation (3) properties.
The current study retrospectively reviewed the clinical course of 10 patients with a diagnosis of HVLLPD who were treated with IVIG in our department from 2008 to 2018. The clinical characteristics of 10 patients (6 males and 4 females), mean age 8.5 years (range 3–24 years), is summarized in (Table SI). Two patients had adult-onset HVLLPD. All the patients had extracutaneous manifestations. All the proliferating lymphocytes were T-cell type. One patient was lost to follow-up.
IVIG was used in all patients, at a dosage of 0.4 g/kg 3–5 days monthly. After the disease stabilized, IVIG was reduced gradually every month for 3 months. Stable disease was defined as less than 5 relapses in a year. Short-term systemic glucocorticoids were administered when large ulcers were present or high fever persisted for over 3 days. Glucocorticoids were tapered quickly after episodes faded. Initially, 8 of the 10 patients received glucocorticoids. The maximum dose was prednisone 30–40 mg/day and the duration was 1–2 weeks. After IVIG therapy, only 3 of the 7 patients in remission still needed glucocorticosteroids to control the episode. However, both the dose and duration were reduced.
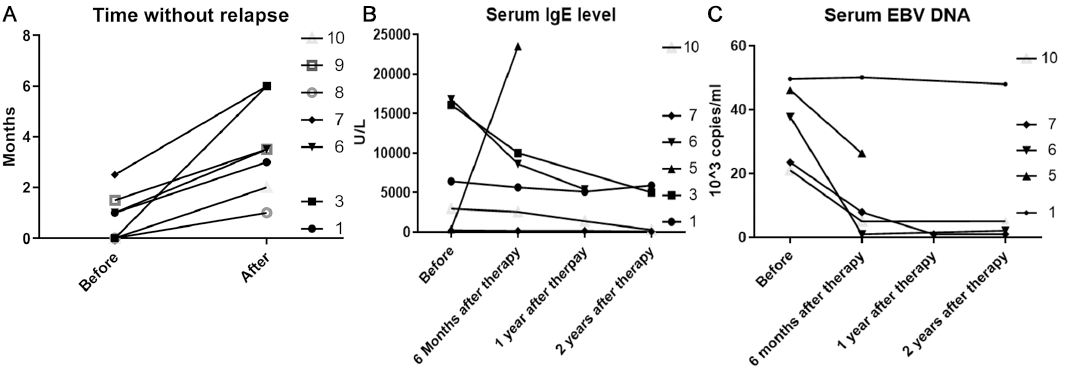
The survival rate was 8/9 (mean follow-up 38.2 months). Progression-free survival was 36.7 months. Seven patients demonstrated improvement with IVIG: the time without relapse was prolonged (Fig. 1A). Compared with initial symptoms, extracutaneous symptoms ameliorated during the follow-up (Table SI). Splenomegaly resolved in 3 of the 6 patients. Fever and lymphadenopathy resolved in 3 of the 7 patients. Hepatomegaly resolved in 1 patient. A decrease in IgE level was observed in 4 patients and a decrease in EBV-DNA level in 4 patients (Fig. 1B, C). No change in blood eosinophil count was observed. No severe systemic viral or bacterial infection was observed. One patient (case 4) died of pancytopaenia after being referred to the paedi-atric department where she received chemotherapy. One patient (case 5) progressed to NK-T-cell lymphoma.
Fig. 1. (A) The time (months) between episodes before and after intravenous immunoglobulin (IVIG). (B): Serum immunoglobulin (Ig)E levels before, 6 months, 1 year and 2 years after IVIG. (C) Epstein–Barr virus (EBV)-DNA before, 6 months, 1 year and 2 years after IVIG.
In the literature, although 15% of cases of HVLLPD may develop to systemic lymphoma (4), most patients have an indolent course. In some patients, the disease may persist for over 25 years (5). Different studies had heterogeneous results about death rate, varying from 0–100% (6), which indicated that HVLLPD could represent a large spectrum of clinical patterns and therefore it is difficult to predict the prognosis. In the Chinese population, the death rate varies from 20–47.4% (7, 8). The main cause of death is systemic lymphoma, hepatic failure and complications secondary to chemotherapy. HSCT, chemotherapy, glucocorticoids, cyclosporine, interferon α, thalidomide and minocycline have been reported to show success in treating HVLLPD. Although HSCT may be curative for HVLLPD, the lengthy waiting time before transplantation, the unpredictable risk of HSCT, and the high cost makes it difficult to implement. Chemotherapy may induce short-term remission, but may result in progression and rapid relapse of disease or severe side-effects. Several studies have shown that chemotherapy may be associated with poor prognosis. Immunosuppressors, immunomodulators and IFN-γ may be effective in some patients, but their efficacy needs to be proved by more cases.
Although the pathogenesis of HVLLPD remains unknown, the over-proliferation of lymphocytes caused by repeated activation of the virus may be the key components (9). Given the nature of HVLLPD and the limitations of current therapies, there remains a need for a less aggressive, but efficient therapy. IVIG seems to be a safe and ideal choice because of its anti-viral and anti-inflammation characteristics. The anti-viral activity of IVIG is mediated by different specific antibodies to different pathogens, including EBV (10). The anti-inflammatory function is mediated by Fc fragment sialylation, inhibiting CD8+ T cell, modulating NK cell and inhibiting NF-κB pathway. In practice, IVIG (11) had been used in the post-transplant lymphoproliferative disorders, as IVIG could increase antibody against Epstein-Barr nuclear antigens and reduce Epstein-Barr viral load. Lastly, patients with HVLLPD are prone to infections and often present with hyper-IgE serum level (7). IVIG can help patients with HVLPPD reduce the risk of infection and avoid complications. Thus, IVIG therapy could be effective in treating HVLLPD.
These are the first reported cases of IVIG as treatment for HVLLPD. A review of our patients showed that the death rate is lower than previously reported and the hospitalization costs for our patient are reduced, as no extra antibiotics or therapy secondary to chemotherapy were required. Thus, IVIG may be a cost-effective, safe therapy for HVLLPD, which, as demonstrated by our study, can be associated with a higher survival rate, and a significant decrease in both severity and frequency of HVLLPD.


 Click to show fullsize
Click to show fullsize