


1Department of Dermatology, 2Department of Pediatrics, Takamatsu Red Cross Hospital, 4-1-3 Ban-cho, Takamatsu, Kagawa 760-0017, 3Department of Dermatology, Faculty of Medicine, Institute of Medical Pharmaceutical and Health Science, Kanazawa University, Kanazawa, and 4Department of Dermatology, Osaka University Graduate School of Medicine, Suita, Japan. *E-mail: t-hamada@cc.okayama-u.ac.jp
Accepted Jul 7, 2020; Epub ahead of print Jul 29, 2020
Acta Derm Venereol 2020; 100: adv00238.
Juvenile dermatomyositis (JDM) is a rare autoimmune condition that affects skeletal muscle and skin with onset in childhood. The characteristic presentation of JDM includes proximal muscle weakness and a disease-specific skin rash including a heliotrope rash, V-neck sign, shawl sign and Gottron’s papules.
DM autoantibodies may be myositis-specific (MSAs) or myositis-associated (MMAs) autoantibodies, the latter of which are detected in patients with overlap features of idiopathic myositis and other connective tissue diseases (1). In recent years, individual MSAs have been proven to be relevant factors in the distinct disease phenotype of DM (2).
We report here the very rare case of a Japanese child with DM, who presented with the typical skin rash and an absence of clinical muscle weakness, suggesting anti-Tif1γ DM. The patient’s sera reacted with both Tif1γ and Mi-2.
A 2-year-old Japanese girl experienced erythema and papules on her fingers and knees. One month later, facial erythema occurred after exposure to sunlight. She had been treated with a topical corticosteroid, resulting in a refractory clinical course. Four months later, the patient was referred to us because of her progressing skin rash.
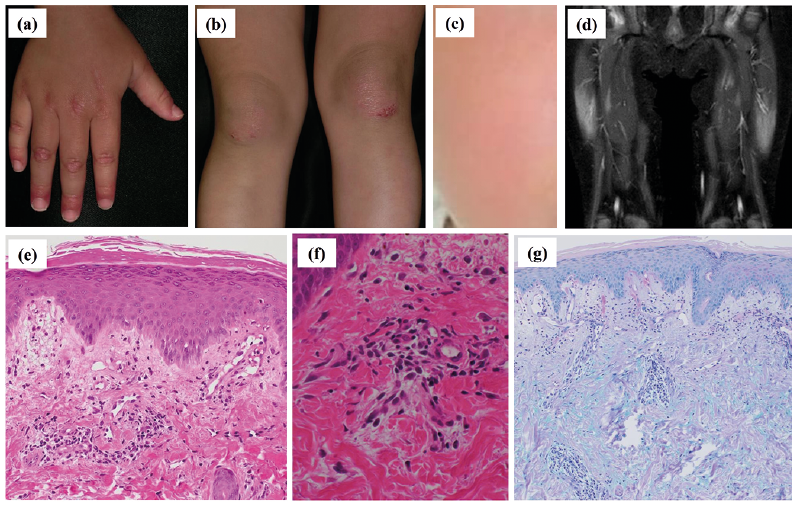
A physical examination revealed typical skin rash on the face, hands, fingers and knees (Fig. 1a–c). No myalgia was clinically observed. Routine laboratory tests revealed a white blood cell count of 8,570/µl, increased serum levels of creatinine kinase at 467 U/l (43–290) and aldolase at 13.5 U/l (2.7–7.5). Circulating autoantibodies against the nucleus were detected at a titre of 1:320, with a speckled pattern by indirect fluorescent antinuclear antibody testing. No active lung involvement was detected by auscultation or chest X-ray imaging. Inflammation of the bilateral quadriceps muscles was detected by magnetic resonance imaging (Fig. 1d). No malignancy was observed. A biopsy specimen from an erythema on the knee showed dermal perivascular lymphocytic infiltrates and marked oedema of the superficial dermis with mucin deposits (Fig. 1e–g). No muscle weakness was observed by manual muscle testing.
Fig. 1. Clinical, histopathological and magnetic resonance imaging (MRI) findings. (a, b) Periungual erythema of the fingers, erythemic papules on the extensor side of the interphalangeal joints of the hands, and infiltrating erythemic plaques on the knees. (c) Facial erythema on the cheek. (d) Magnetic resonance imaging shows inflammation of the bilateral quadriceps muscles (short TI inversion recovery image). (e–f) Dermal perivascular lymphocytic infiltrates and edema of the superficial dermis with mucin deposits. (Haematoxylin and eosin stain: original magnification (e) ×100, (f) ×400; Alcian blue stain: (g) ×40).
Enzyme-linked immunosorbent assays (ELISAs) for MSAs were positive for Tif1γ and Mi-2 at titres of 60 (cut-off < 32) and 54 (cut-off < 53), respectively. In addition, the patient’s sera reacted with Tif1γ and Mi-2 in immunoprecipitation-Western blotting analysis, carried out as described by Kang et al. (3) (Fig. S1). Taken together, a diagnosis of clinically amyopathic JDM with autoantibodies to both Tif1γ and Mi-2 was made.
The patient was initially treated with 2 rounds of intravenous methylprednisolone pulse therapy (30 mg/kg daily, for 3 consecutive days) followed by oral prednisolone at a dose of 1.0 mg/kg daily and oral methotrexate at 5 mg/m2 weekly (Fig. S2). Her skin rash resolved quickly and her muscle enzyme serum levels normalized. Her eruption recurred when oral prednisolone was tapered to 0.6 mg/kg daily. Twelve months after therapy was initiated, the patient had no myositis with oral prednisolone at 0.2 mg/kg daily and methotrexate at 15 mg/m2 weekly, despite a persistent course of skin rash on the face, knees and fingers (Fig. S2). Physical examination revealed no abnormal findings suggestive of malignancy. Her serum levels of anti-Tif1γ and anti-Mi-2 autoantibodies were continuously elevated at 87 and 55, respectively.
JDM is a rare inflammatory myopathy, mainly involving proximal skeletal muscle and with a disease-specific skin rash. The mean age of onset is 6.7 years in boys and 7.3 years in girls, with a female to male ratio of 2.3 (4). Unlike adult DM, no internal malignancy is generally associated with JDM (5).
To date, several MSAs have been identified in patients with idiopathic inflammatory myositis, including DM. Each MSA affects the clinical characteristics and prognosis in cases of both JDM and adult DM (6). Patients with anti-Mi-2 autoantibodies show classical DM with the typical skin rash and proximal muscle weakness, and serum levels of creatinine kinase are more likely to be high compared with those in patients with other MSAs or MAAs (7). Anti-Mi-2 DM patients respond well to systemic therapies including corticosteroids, with a favourable clinical course (8).
Anti-Tif1γ autoantibodies are detected in 22–35% of JDM cases (1, 6, 9). The presence of circulating anti-Tif1γ autoantibodies is a powerful predictor for malignancy in adult DM (10). In very rare cases, paediatric patients with DM may develop malignancy within a mean time of 12 months after the onset of DM (11). Juvenile and adult patients with anti-Tif1γ autoantibodies tend to have typical, severe skin rash, including heliotrope rash, V-neck sign, Shawl sign and Gottron’s papules with a chronic clinical course (6, 12). Extensive photo-distributed erythema and occasional dysphagia may occur, with fewer systemic symptoms than are seen in other types of DM (12). The current patient showed extensive skin rash, despite less muscle weakness, which matches the typical clinical features of anti-Tif1γ DM.
The sera of the current patient reacted with both TIF1γ and Mi-2 by ELISA and immunoprecipitation-Western blotting. Occasionally, weak reactivity to anti-Tif1γ autoantibody may be detected in adult patients with anti-Mi-2 polymyositis (PM)/DM. Anti-Mi-2 autoantibody may cross-react with Tif1γ protein via a plant homeodomain (PHD) that is highly homologous to a PHD of Mi-2β protein (13). Thus, the positive result for both anti-TIF1γ and anti-Mi-2 autoantibodies in the current patient’s sera may have been caused by this cross-reaction.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize