


1St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust, London, 2Department of Dermatology, Brighton and Sussex University Hospitals NHS Trust, Brighton, UK, 3Department of Pathology, Instituto Português de Oncologia de Lisboa, Lisboa, Portugal, 4Department of Plastic Surgery, Guy’s and St Thomas’ NHS Foundation Trust, 5Genetic Skin Disease Group, King’s College London and 6Department of Dermatology, Great Ormond Street Hospital for Children NHS Foundation Trust, London, UK
Epidermolysis bullosa (EB), notably severe recessive dystrophic EB (RDEB-S), is associated with increased risk of aggressive mucocutaneous squamous cell carcinomas, the major cause of mortality in early adulthood. This observational, retrospective case review describes a series of EB patients with cutaneous squamous cell carcinomas over a 28-year period. Forty-four EB patients with squamous cell carcinomas were identified with a total of 221 primary tumours. They comprised: 31 (70%) with RDEB-S, 4 (9%) with other RDEB subtypes, 5 (11.4%) with dominant dystrophic EB, 3 (6.8%) with intermediate junctional EB and 1 (2.3%) with Kindler EB. Squamous cell carcinomas occurred earlier in RDEB-S (median age 29.5 years; age range 13–52 years) than other groups collectively (median age 47.1 years; age range 30–89 years) and most had multiple tumours (mean 5.8; range 1–44). Squamous cell carcinoma-associated mortality was high in RDEB-S (64.5%), with median survival after first squamous cell carcinoma of 2.4 years (range 0.5–12.6 years), significantly lower than previous reports, highlighting the need for early surveillance and better treatments.
Key words: epidermolysis bullosa; squamous cell carcinoma; cancer; prognosis.
Accepted Jul 6, 2021; Epub ahead of print Jul 7, 2021
Acta Derm Venereol 2021; 101: adv00523.
doi: 10.2340/00015555-3875
Corr: Jemima E. Mellerio, St John’s Institute of Dermatology, Guy’s and St Thomas’ NHS Foundation Trust, Westminster Bridge Road, London SE1 7EH, London, UK. E-mail: Jemima.mellerio@kcl.ac.uk
This series is the largest reported to date and highlights that individuals with different forms of epidermolysis bullosa are at risk of developing squamous cell carcinomas of the skin. Patients often develop multiple primary tumours that tend to behave aggressively despite wide local excision and have poor response to conventional chemotherapy. The prognosis in severe recessive dystrophic epidermolysis bullosa is worse than previously reported, with median survival from diagnosis of first squamous cell carcinoma of only 2.4 years.
Epidermolysis bullosa (EB) encompasses a group of rare inherited skin fragility disorders caused by mutations in genes encoding dermal-epidermal adhesion proteins (1). Four main types of EB are recognized, determined by the level of skin cleavage; EB simplex (EBS), junctional EB (JEB), dystrophic EB (DEB) and Kindler EB (KEB). These clinically heterogeneous entities have multiple phenotypic subtypes (2). Some forms are associated with cutaneous and mucosal squamous cell carcinoma (SCC). Unlike cutaneous SCCs in the general population, where chronic ultraviolet radiation (UVR) exposure predisposes to tumours on sun-exposed sites, EB-associated SCCs arise at sites of chronic skin blistering, wounds and scarring (3–6). In addition, in EB, multiple primary SCCs often develop, and tumours behave aggressively with high risk of metastasis. EB SCCs are the leading cause of death in individuals with severe recessive DEB (RDEB-S) (3).
The tumour micro-environment is critical for initiation, progression and subsequent metastatic potential of EB SCCs (for review see (7)). Whilst RDEB SCCs harbour mutations in many of the same driver genes as non-EB SCCs (e.g. NOTCH1, NOTCH2, TP53, CDKN2A and HRAS), UVR signature mutations are not seen. High levels of mutations driven by the APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) family of enzymes are observed in RDEB SCCs and raised levels of certain APOBEC members are found in chronically inflamed RDEB skin (8). Loss of type VII collagen in RDEB contributes to the profibrotic dermal state, partly through transforming growth factor (TGF-β) activation, which increases the stiffness of the extracellular matrix, creating a permissive tumour microenvironment (9, 10). Unbiased proteomic analyses of RDEB SCCs have also highlighted the role of tissue damage, extracellular remodelling and bacterial challenge in the development of these tumours (11). In addition, RDEB fibroblasts demonstrate a cancer-associated fibroblast-like profile leading to a favourable environment for SCC development (12). The unrelenting inflammation observed in RDEB skin may also influence tumourigenesis. Interleukin-6 (IL-6), another cytokine that correlates with EB disease severity, may be involved in skin fibrosis, tumour growth and metastatic potential in EB SCCs (13, 14). Microbial colonization and infection are possible carcinogenesis drivers in EB (15). Recent evidence also supports a defect of innate immunity in RDEB (16). Moreover, upregulation of complement components in RDEB SCCs may also contribute to the aggressive nature of these cancers (17).
Most epidemiological data on EB SCCs have emerged from the USA National EB Registry which showed an increased cumulative risk of developing SCC in EB with age. In RDEB-S, the cumulative risk of developing at least one SCC was 7.5% by age 20 years, 67.8% by 35 years, and 90.1% by 55 years, paralleled by an increased cumulative risk of SCC-related death of 38.7% by age 35 years, 70.0% by 45 years, and 78.7% by 55 years (3). Other series have found similar outcomes, namely early-onset tumours in RDEB-S and median survival after first SCC of between 4 and 11 years (4, 5). The objectives of the current study were to review all SCCs presenting in patients with EB seen at the London EB centres, and to report the experience and management of these tumours over 28 years.
A retrospective case review was conducted of all patients with EB treated at the London EB centres (Guy’s and St Thomas’ NHS Foundation Trust and Great Ormond Street Hospital for Children NHS Foundation Trust) diagnosed with SCC between 1 July 1991 and 30 June 2019. Local ethical approval was not required for this retrospective study.
The onset of each primary SCC was taken as the time of the patient’s presentation to our services. Tumours occurring at the site of previous primary SCCs or at previous excision margins were considered to be recurrences unless arising more than 12 months after excision of the initial primary at that site.
All patients had the diagnosis of EB type and subtype confirmed on skin biopsy findings (direct immunohistochemistry for expression of relevant basement membrane zone proteins and/or transmission electron microscopy) and/or genetic testing, clinical features and family history.
Epidermolysis bullosa subtype
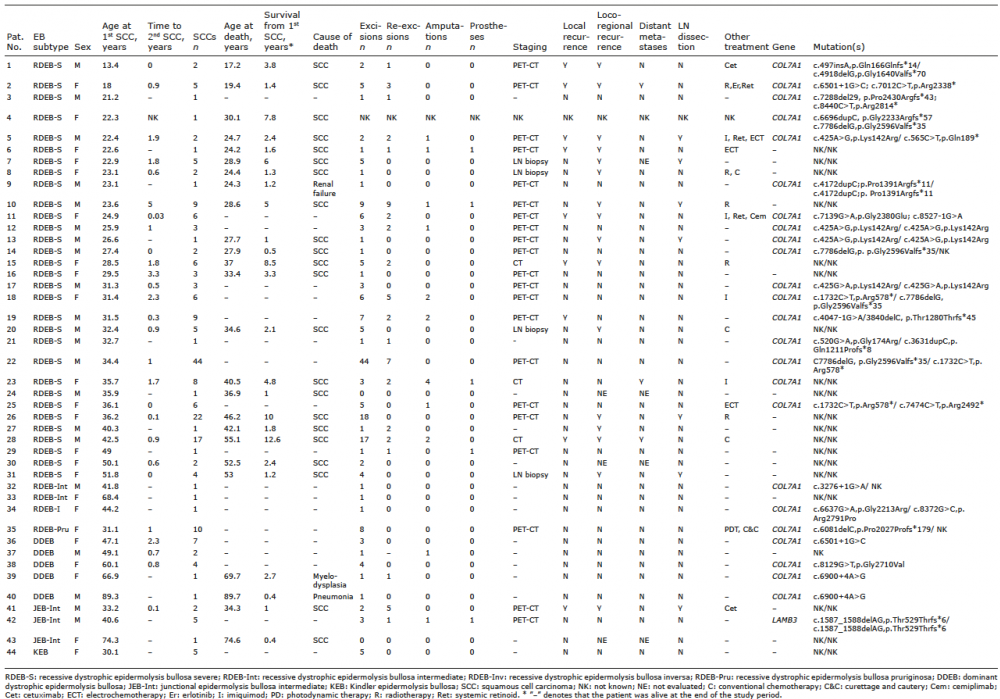
Forty-four patients had a cutaneous SCC during the study period (Tables I and II). EB subtypes included: 31 (70%) with RDEB-S, 2 (4.5%) with intermediate RDEB (RDEB-Int), 1 (2.3%) with RDEB-pruriginosa (RDEB-Pru), 1 (2.3%) with RDEB-inversa (RDEB-Inv), 5 (11.4%) with dominant DEB (DDEB), 3 (6.8%) with intermediate JEB (JEB-Int) and 1 (2.3%) with KEB. No mucosal SCCs were identified in the study cohort during the period of data collection.
Table I. Individual patient data for epidermolysis bullosa (EB) subtype, timing of squamous cell carcinomas (SCC(s)), mortality, treatments and genotype
Table II. Summary of patient SCC data by epidermolysis bullosa subtype
Age at first squamous cell carcinoma
The total median age at first SCC was 32.8 years (age range 13–89 years) but was earlier in RDEB-S with median age of 29.5 years (age range 13–52 years) (Table I).
Number of squamous cell carcinomas
In total, 221 primary SCCs occurred in the 44 patients, of whom 28 had multiple tumours. The median number of tumours per patient was 2.5 (range 1–44). Twenty-two (71%) of the RDEB-S cohort had multiple primaries, with a median of 3 tumours each (range 1–44). Patients with JEB-Int had a median of 2 tumours each (range 1–5), as did those with DDEB (range 1–7). Individuals with RDEB-Pru and KEB had 10 and 5 tumours, respectively (Table I).
Anatomical sites affected
Most SCCs occurred on the limbs, with tumour distribution differing between EB subtypes (Fig. 1). No tumours arose on the head or neck, or at mucosal sites.
Fig. 1. Anatomical distribution of primary squamous cell carcinomas arising in patients with different forms of epidermolysis bullosa (EB). Most severe recessive dystrophic EB (RDEB-S) tumours arose over bony prominences, particularly the joints of hands and feet. Dominant dystrophic EB (DDEB) and RDEB- pruriginosa (RDEB-Pru) tumours were more common on the lower legs, particularly the shins. All squamous cell carcinomas in the Kindler EB (KEB) patient arose on the hand. JEB: junctional EB; Int: intermediate; Inv: inversa.
Locally recurrent disease
Eight patients exhibited local recurrence within 12 months (median 4, range 1–7 months) of having moderately (n = 2) and poorly differentiated SCCs (n = 6) completely excised. One patient (Patient 1) developed simultaneous local recurrence with widespread subcutaneous metastases on the same limb 4 years after complete excision of 2 poorly differentiated primary SCCs.
Metastatic disease
Seventeen patients had metastatic disease detected on 18-fluorodeoxyglucose positron emission tomography/computed tomography (18-FDG PET/CT), plain CT staging and/or lymph node biopsy/dissection. Of these, 16 (94%) had died by the end of the study period.
Tumour intervals
The median time between first and second SCCs in RDEB-S was 0.9 years (range 0–3.3 years). Of these patients, 2 presented with 2 synchronous primaries, one had 3 synchronous primaries and another patient, 4 synchronous primaries. A further 2 patients developed their second SCC within one month of the first. In patients with multiple tumours, the interval between developing subsequent SCCs tended to decrease over time, with a crescendo of tumour development leading to death (Fig. 2). Increased multifocal disease was also observed over time.
Two JEB-Int patients had multiple primary SCCs with intervals between the first and second tumours of 1 month and 13.7 years, respectively. Three DDEB patients had multiple primary SCCs with a median interval between first and second tumours of 0.8 years (range 0.7–2.3 years).
Fig. 2. Temporal occurrence of squamous cell carcinomas (SCCs) in each patient with severe recessive dystrophic epidermolysis bullosa. Each patient is represented as a horizontal line showing the occurrence of their tumours from the age at first tumour denoted on the y-axis. The size of the circle denotes the number of SCCs diagnosed at each time point. In some patients there was a trend for developing greater numbers of tumours at shorter intervals over time.
Histological characteristics/tumour differentiation
Most of the 221 SCCs were well-differentiated (53.4%), followed by moderately-differentiated (29.4%), in situ (9.5%) and poorly-differentiated (7.7%). In 16 (36%) patients (13 RDEB-S, 2 JEB-Int, 1 RDEB-Pru; patients 1, 2, 5, 10, 11, 14, 15, 17, 18, 20, 25, 28, 30, 35, 42, 43) there was a trend over time of change from well- to poor-differentiation of subsequent SCCs (including recurrences) and in 11 of these 16 (69%) patients, this was associated with subsequent mortality. There were 5 patients (4 RDEB-S and 1 JEB-Int; patients 6, 9, 14, 16, 41) with poorly-differentiated initial primary SCCs, leading to poor outcomes with median survival of only 1.2 years (range 0.5–3.9 years). However, 2 RDEB-S patients developed metastatic disease after having only well-differentiated SCCs completely removed (patients 26 and 31) and a further 2 RDEB-S patients with completely excised moderately-differentiated primary SCCs developed metastatic disease 4 months and 4 years later, respectively (patients 1 and 13). One RDEB-S patient (patient 22) had 44 primary SCCs (ranging from in situ to poorly differentiated) completely excised and had no evidence of metastatic disease 15 years after initial SCC diagnosis.
Squamous cell carcinoma management
Wide local excision. Most SCCs (191/221; 86.4%) underwent wide local excision (WLE). Thirty-five (18.3%) of these had re-excision for incomplete margins. No patients underwent Mohs micrographic surgery.
Therapeutic lymphadenectomy. Six patients (5 RDEB-S, 1 JEB-Int) had 7 therapeutic lymphadenectomies for clearance of regional metastatic disease (4 axillary and 3 inguinal/pelvic). In all, surgery was well-tolerated with good wound healing. Median survival post-operatively was 0.86 years (range 0.2–2.6 years). No patients underwent sentinel lymph node biopsy.
Inoperable disease. Two patients had inoperable disease at diagnosis due to large multi-focal SCCs (one with RDEB-S, patient 14, and one with JEB-Int, patient 43). Both opted for palliative care and died 6 and 5 months after diagnosis, respectively.
Radiotherapy. Five RDEB-S patients received palliative radiotherapy. In 4, this was for symptom control of ulcerated metastatic disease; all showed slowed tumour growth and improved wound healing following 45–50 Gy delivered over 20–25 fractions. One received treatment for symptom control of bony metastases with 20 Gy delivered over 5 fractions, but did not respond.
Conventional chemotherapy. Three RDEB-S patients with metastatic disease commenced conventional chemotherapy with 5-fluorouracil and cisplatin; however, disease progressed in all cases.
Electrochemotherapy. Two RDEB-S patients received bleomycin electrochemotherapy for locally aggressive disease. No significant tumour response occurred, and both reported significant pain. One developed severe post-operative sepsis.
Targeted biologic therapy. Two patients (1 RDEB-S, 1 JEB-Int) received palliative treatment with intravenous (i.v.) epidermal growth factor receptor (EGFR) inhibitor, cetuximab. Tumour expression of EGFR was not checked in either case. Treatment was well-tolerated in both and 1 reported improved pain control. However, due to disease progression, treatment was discontinued after 3 and 7 weeks, respectively.
One RDEB-S patient received oral tyrosine kinase inhibitor, erlotinib. Treatment was discontinued due to disease progression and adverse effects, including acne and diarrhoea.
A further RDEB-S patient with metastatic SCC received 3-weekly i.v. cycles of programmed-cell death receptor (PD-1) inhibitor, cemiplimab. There was reduced tumour size, improved local symptoms (pain, odour and exudate) and she was alive 6 months after starting therapy at the end of the study period.
Topical treatments. Imiquimod 5% cream was used in 7 patients. Treatment was applied 2–3 times weekly for up to 12 weeks or until an inflammatory response was reached. In 4 patients (2 RDEB-S, 1 JEB-Int, 1 RDEB-Int) in situ SCC was successfully cleared. Another RDEB-S patient with in situ SCC disease did not respond; the lesion was subsequently excised and found to be a well-differentiated SCC. Imiquimod was trialled in 1 RDEB-S patient with invasive SCC to reduce tumour size pre-operatively, without response. Palliative imiquimod treatment was used for a further RDEB-S patient with a large fungating tumour on the hand. Although the tumour shrank, treatment was not tolerated due to severe inflammation and pain.
One patient with RDEB-Pru had photodynamic therapy (PDT) for in situ SCC on the lower leg. Treatment was well tolerated, although complicated by post-operative skin infection, ulceration and persistent disease. She subsequently had successful serial curettage and cautery.
Systemic retinoids. After first SCC diagnosis, one RDEB-S patient (patient 5) started acitretin 10 mg daily which was well-tolerated apart from hair loss. Unfortunately, further SCCs developed despite continuing acitretin for 2 years. Two other female RDEB-S patients (patients 2 and 11) received isotretinoin 10 mg daily. Both discontinued after 1 month due to dry eyes in 1 and deteriorating liver function in the other.
Declined treatment. One RDEB-S patient (patient 4) declined further care after excision of her primary SCC and reportedly died 11.8 years later from metastatic disease. A second RDEB-S patient (patient 27) had 3 incomplete excisions for SCC at his local hospital, then declined all further interventions and died 1.8 years later from metastatic SCC. Another RDEB-S patient (patient 24) declined intervention for SCC after diagnostic punch biopsy and died 1 year later from metastatic disease. A further RDEB-S patient (patient 16) declined intervention for her second primary SCC, dying 8 months later.
Amputation and prostheses. Twelve patients (27.3%) required a total of 17 amputations (10 RDEB-S, 1 DDEB, 1 JEB-Int). Twelve amputations cleared the SCC (5 below-knee amputations (BKA), 1 above-knee amputation (AKA), 1 forearm amputation, 5 finger amputations). Palliative amputations were also performed for locally aggressive disease (1 hand and 1 AKA).
One RDEB-S patient (patient 23) was offered right hand amputation for multifocal SCC, which, however, was declined as this was her dominant hand. She had 3 more years living independently until she required palliative symptomatic forearm amputation. A hand prosthesis was not tolerated due to discomfort.
Another RDEB-S patient (patient 19) was offered right proximal forearm amputation for persistent hand SCC following multiple WLE attempts. He similarly declined as this was his dominant hand. After 18 months, hand amputation was performed for symptom control, followed by amputation of the forearm stump due to progressive disease.
One RDEB-S patient who had BKA (patient 10) and another with JEB-Int (patient 42) who underwent AKA were fitted for leg prostheses with good effect, both enabling independent ambulation. The 4 remaining BKA patients (1 DDEB; patient 37), (3 RDEB-S; patients 12, 23, 25) used wheelchairs pre-amputation and did not require prostheses.
Outcomes
At the end of the study period, 25 (56.8%) patients had died, 22 of these (88%) from SCC. Others died from unrelated causes; 1 with DDEB (patient 40) from pneumonia aged 90 years, another with DDEB (patient 39) from myelodysplasia at 70 years, and 1 with RDEB-S (patient 9) from renal failure aged 24 years. The majority of the 22 deaths from SCC had RDEB-S (n = 20), the remainder JEB-Int (n = 2).
RDEB-S patients died from SCC earlier, with a median age of death of 31.8 years (range 17–55 years) compared with 54.4 years (range 34–74 years) for JEB-Int (Table I). These RDEB-S patients had a median of 2.5 SCCs by time of death, but the range was wide (1–22 primaries).
The median survival from diagnosis of first SCC in RDEB-S was 2.4 years (range 0.5–12.6 years) (Fig. 3). The median survival in the 2 JEB-Int patients was 0.7 years (range 0.4–1 years); the first presented late with inoperable disease and the other had rapidly progressive SCC.
In the current series, the median follow-up of survivors to the end of the data collection period was 3.9 years (range 1.1–16.4 years) in individuals with RDEB-S and 7.2 years (range 1.5–26.7 years) in all other subtypes combined.
Fig. 3. Overall survival from diagnosis of first squamous cell carcinomas in patients with severe recessive dystrophic epidermolysis bullosa.
Our case series is the largest reported to date, with 221 primary SCCs arising in 44 well-characterized patients with different EB subtypes. It provides further insight into the natural history of these tumours and information on different treatments for local disease and distant spread.
Anatomical location
In keeping with previous reports (4–6), most SCCs arose on the limbs, particularly over the hands, feet and shins. This correlates with areas most prone to chronic ulceration, scarring and infection, supportive of the role of bacterial colonization, a fibrotic stroma and increased inflammation in driving tumour initiation and progression. This largely photo-protected distribution supports the lack of UVR in EB SCC pathogenesis.
Epidermolysis bullosa subtype and epidemiology
Consistent with previous studies, this study shows that patients with RDEB-S are most at risk of developing SCC, but milder subtypes are also susceptible (1–4, 18). Thirteen of our cohort (30%) had non-RDEB-S phenotypes, underscoring the need for surveillance in JEB-Int, KEB and other DEB patients, albeit with tumours generally arising later.
EB-associated SCCs occur early in life, particularly in RDEB-S. The youngest patient in our series was 13.4 years at first SCC, although this has been described in a child of just 6 years (19). The median age of SCC onset in our RDEB-S cohort was 29.5 years, compared with 23 years in another series (5). As our EB database does not date from the start of our data collection, we were unable to calculate cumulative risks of developing SCCs. However, previous data from the National EB Registry (NEBR) in the USA delineated a cumulative risk of developing a first SCC in RDEB-S of 7.5%, 52%, 80% and 90% by age 20, 30, 45 and 55 years, respectively (3), mirrored by similar figures in Australasia of 26% and 64% by ages 20 and 30 years, respectively (5).
Interestingly, the current study found a median survival following diagnosis of first SCC in RDEB-S of 2.4 years (range 0.5–12.6 years), less than the previously reported median survival of 4 years in the Australasian series, which included only 11 RDEB-S patients compared with our 31 (5). Castelo et al. (6) found a median survival of 11.4 years after first tumour, in their series of 13 RDEB patients without specified subtype, and 1 with KEB, invalidating comparison with just RDEB-S patients. In the NEBR cohort, it was noted that most RDEB-S patients died within 5 years of diagnosis of first tumour, with no median interval given (3).
Due to aggressive disease in our JEB-Int cohort (n = 2), the median survival of 0.7 years (range 0.4–1 years) was shorter than the reported mean of 8.9 years (18). The median age of first SCC in our JEB patients was 40.6 years, compared with 52 years in the literature, where multiple primaries were also frequent (in 9 of 14 patients reviewed) (18).
In patients with multiple primary SCCs in our series, there tended to be an acceleration in the numbers of tumours developing over time, which, to our knowledge, has not been documented previously. Interestingly, not all patients with multiple primaries have an increasingly aggressive course. Patient 22 with RDEB-S who had 44 primaries remained alive with no metastatic spread 15 years after his first primary; the reasons behind the apparently different biological course of his disease remains elusive and unexplained by his COL7A1 genotype.
Squamous cell carcinoma treatment
WLE was performed in most SCCs in the current series, consistent with recommended best practice (20). In 35/191 (18.3%) excisions, however, margins were incomplete, underscoring difficulties in delineating tumours on a background of ulcerated and scarred EB skin. In approximately a quarter of patients, when WLE was not achievable due to tumour size or anatomical constraints, amputation was undertaken, taking into account patients’ preferences. This figure is similar to previously reported rates (4). Two patients tolerated prostheses supporting previous reports of their use in EB (21).
Contrary to previous reports (22, 23), radiotherapy was tolerated well in 5 of our patients, presumably because of delivery through smaller, more numerous fractions. It was only used palliatively and successfully controlled symptoms in most patients.
Conventional chemotherapy was used in 4 patients, each with poor response, supporting recommendations that risks may outweigh potential benefits (20). Our experience with cetuximab was equally disappointing, despite promising reports in the literature (24–28). Cemiplimab, a PD-1 inhibitor approved by the Food and Drug Administration and European Medicines Agency for the treatment of advanced cutaneous SCC, was administered to one of our cohort who had partial tumour regression. This, and other PD-1 inhibitors such as nivolumab and pembrolizumab, both approved for the management of advanced head and neck SCC, may have a role in EB SCC palliation (26, 28–31). Following in vitro work in EB SCC cell lines and an in vivo mouse model (32), a clinical trial of the polo-like kinase inhibitor rigosertib is currently underway (ClinicalTrials.gov: NCT01807546). Recent preclinical studies have also identified the JAK1/2 inhibitor, ruxolitinib (33), and TGF-β receptor 1 kinase inhibitors (34) as potential therapies for RDEB-associated SCCs.
Adjunctive topical therapies may be considered in EB SCC. PDT has been used successfully in a single case of EB for in situ disease (35). Although tolerable, the treatment was unsuccessful for our patient. In the current series imiquimod was successful and well-tolerated for in situ SCC in 4 patients.
Oral retinoids have been of benefit for chemoprevention of SCCs in the organ transplant population (36), but although tolerable in EB (37), any effect on tumourigenesis in EB is not yet established.
Whilst many different treatment modalities have been tried in EB cancers, some individuals in our cohort chose to forgo tumour clearance surgery by limb amputation in favour of living with their SCC to maintain independence. This highlights the need to balance patient preferences, prioritization of function and quality of life with post-intervention survival.
Conclusion
Cutaneous SCCs in individuals with EB occur early and behave aggressively, despite WLE and irrespective of histological grade. They arise at sites of chronic wounds and scarring, particularly on the limbs. This study indicates a worse prognosis in RDEB-S than previous series, with a median survival after a first SCC of only 2.4 years. This reinforces the need for regular skin surveillance in patients with EB, from childhood in RDEB-S and early adulthood in other at-risk subtypes.
Given the rarity of EB-associated SCC, clinical trial data on potential treatments are lacking. However, collaboration between reference centres using different therapies, particularly newer targeted biologic treatments, should yield greater understanding of possible approaches and selection criteria for appropriate patient groups. Ongoing data collection is essential to better understand the pathogenesis of EB SCCs and the reasons for variability in course and prognosis.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize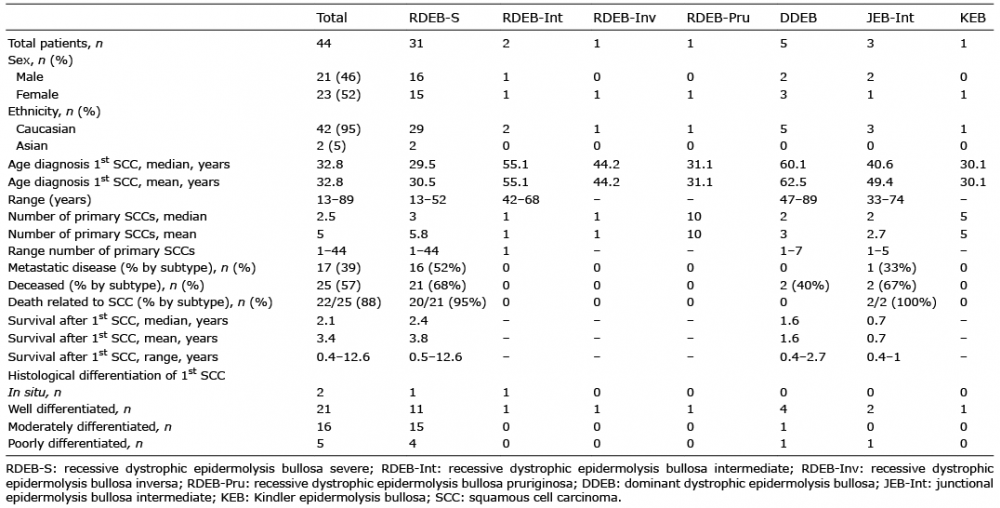 Click to show fullsize
Click to show fullsize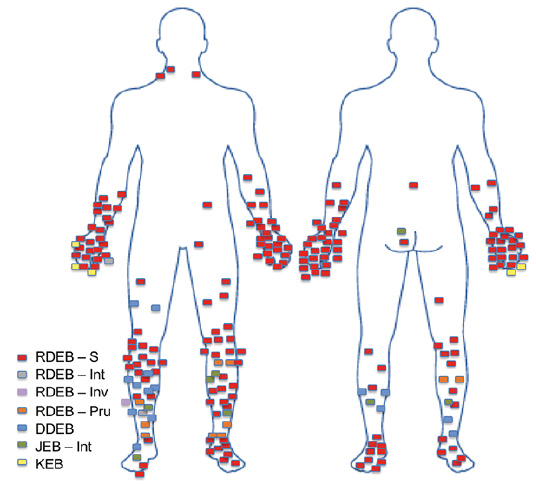 Click to show fullsize
Click to show fullsize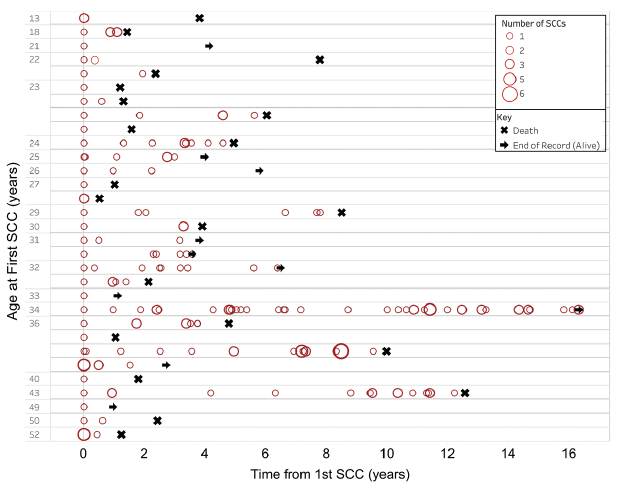 Click to show fullsize
Click to show fullsize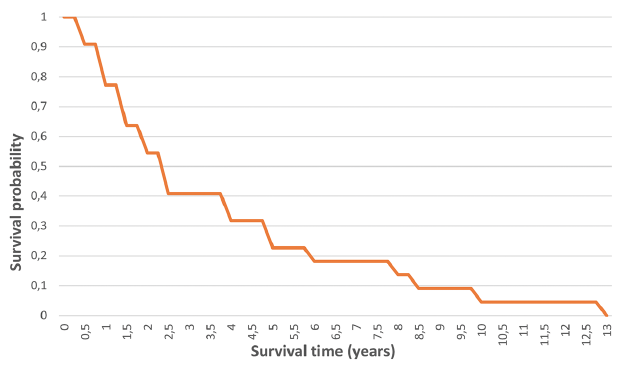 Click to show fullsize
Click to show fullsize