Eosinophilic cellulitis (Wells’ syndrome) is an uncommon inflammatory disease with clinical polymorphism. It is often associated with infectious, allergic or myeloproliferative diseases; however, the exact aetiology is unknown. This report describes a rare case of eosinophilic cellulitis in association with angioimmunoblastic lymphadenopathy. The typical skin findings of Wells‘ syndrome disappeared completely following chemotherapy and autologous stem cell transplantation. Key words: eosinophilic cellulitis; Wells‘ syndrome; flame figures; T-cell lymphoma; AILD.
(Accepted March 29, 2007.)
Acta Derm Venereol 2007; 87: 525–528.
Regina Renner, Ph.-Rosenthalstr. 23–25, DE-04103 Leipzig, Germany. E-mail: Regina.renner@medizin.uni-leipzig.de
Eosinophilic cellulitis was first described by Georg Crichton Wells in 1971 under the name “recurrent granulomatous dermatitis with eosinophilia” (1). In 1979, the name was simplified to the present term “eosinophilic cellulitis” or Wells’ syndrome. It is an uncommon inflammatory dermatosis with suggestive but unspecific histopathological findings and clinical polymorphism. It is often associated with infectious, allergic or myeloproliferative diseases; however, the exact aetiology is unknown. This report is the first to describe a case of eosinophilic cellulitis in association with angioimmunoblastic lymphadenopathy (AILD).
CASE REPORT
Clinical and histopathological findings
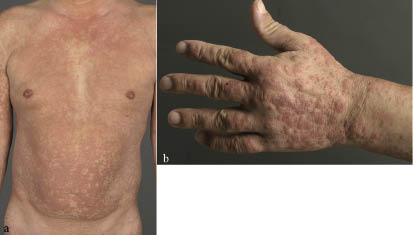
A 48-year-old man was referred to our clinic with recurrent oedema and swelling of the face and neck, which had occurred during the last 3 months. In addition, he reported generalized itching and erythema. Oral antihistamines had been given on suspicion of Quincke’s oedema with chronic urticaria, but they did not control the symptoms. He presented with infiltrated erythemas and multiple livid-erythematous papules and plaques on the integument (Fig. 1a) including the extremities (Fig. 1b) and the soles of his feet. There was generalized lymphadenopathy.
Fig. 1. Infiltrated erythemas and multiple livid-erythematous papules and plaques on (a) the integument and (b) the hand.
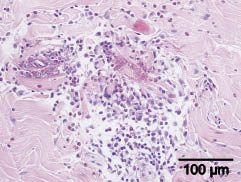
Histopathology from the skin of the thigh showed perivascular and periadnexal as well as diffuse eosinophilic infiltration in the dermis and degenerated basophilic collagen fibres surrounded by eosinophils and eosinophilic granules and histiocytes, the so-called flame figures (Fig. 2). There was no evidence of leukaemic infiltration in the skin.
Fig. 2. Histopathology (H&E ×20) showed perivascular and periadnexal as well as diffuse eosinophilic infiltration in the dermis and the so-called flame figures.
Lymph node biopsy revealed an infiltration of a low malignant non-Hodgkin’s lymphoma (angioimmunoblastic T-cell lymphoma, CD3+, CD5+, CD7-AILD). Bone marrow puncture also showed this infiltration, in addition to a reactive increase in eosinophils. Differential blood count showed eosinophilia of 25.5%. Sonography of the abdomen indicated homogenous splenomegaly. Echocardiography, chest X-ray, otorhinolaryngological control, faecal occult blood test, viral serologies (parvovirus B19, HHV 6, CMV, EBV), Borrelia serology, and syphilis serology showed no significant changes.
Treatment
Therapy was initiated with dapsone 100 mg/day and prednisolone 0.7 mg/kg with decreasing dosage, which led to decreased infiltration and erythema. When the diagnosis of AILD was established, the therapy was switched to chemotherapy. Meanwhile, the patient had received multiple cycles of either vincristine, CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) or ESHAP chemotherapy (etoposide, methylprednisolone, cytarabine and cisplatin). Due to an inadequate response of the AILD to this therapy, autologous stem cell transplantation was carried out. After this procedure, the typical skin findings of Wells’ syndrome disappeared completely, leaving post-inflammatory hypopigmentation.
DISCUSSION
Wells’ syndrome is a chronic recurring inflammatory and pruritic dermatosis of unknown pathogenesis and aetiology. Characteristic, but not specific, are the clinical findings of erythematous-livid plaques, sometimes oedematous or urticaria-like, that may be accompanied by fever, myalgia and (in about 50% of all cases) elevated eosinophilic cells in the blood during the active phase of the disease (1–3). In addition, histological findings show so-called flame figures and further granulomatous infiltration. Our patient showed typical erythematous to urticaria-like plaques accompanied with slight fever, elevated eosinophils in the peripheral blood and histological typical flame figures.
Histopathological features have recently been classified into: (i) an acute stage with oedematous dermis and dense infiltration of eosinophils and histiocytes; (ii) a subacute stage with adherent eosinophils to collagen fibres with degranulation forming the characteristic flame figures as seen in our patient; and (iii) a resolution stage with foreign body type giant cells, histiocytes and few eosinophils (4).
The following diseases or trigger factors have been described to be associated with Wells’ syndrome: insect bites, parasites, viral infections or mycosis, immunization (5), drug eruptions, haemato-oncological diseases or solid neoplasms (6, 7) and TNF-α-inhibitors, such as etanercept or adalimumab (8, 9). Most haemato-oncological diseases associated with eosinophilic cellulitis are mantle cell lymphomas. In our case, triggering factor might have been the additional diagnosed AILD. To our knowledge this is the first time that an AILD associated with Wells’ syndrome has been reported.
A similar course of resolution of the skin lesion following successful treatment of the haematological disease has been described in a case of mantle-zone lymphoma (10).
Wells’ syndrome is thought to be a hypersensitivity reaction to divergent antigens with an eosinophilic reaction (11). Increased interleukin-5 levels have been observed, which mobilizes eosinophils from the bone marrow and facilitates their homing to the skin (12–14). Interleukin (IL)-5 also increases CD25, part of the IL-2 receptor, which promotes degranulation of the eosinophils. This degranulation leads to toxic damage of the tissue and histamine release from basophils (12, 14). Eosinophilic major basic protein (MBP) can be found surrounding collagen fibres (15); this provokes the so-called flame figures. Vasculitis is absent, and direct immunofluorescence findings are negative (3).
The lack of specific or unique symptoms, and an association with different diseases, has formerly led to the identity of Wells’ syndrome being doubted. Aberer et al. (16) postulated it as a distinctive clinical and histological reaction and not just an unspecific symptom of different diseases (17). In contrast, other authors (18) postulated that Wells’ syndrome may be an exclusive cutaneous manifestation of hypereosinophilic syndrome (HES). There are, however, some factors that cast doubt on this hypothesis. In HES, an elevated serum level of MBP can be observed, but there is a lack of typical flame figures, maybe because there is insufficient degranulation of eosinophils in the skin. The pathology of skin lesion in HES is more non-specific than in Wells’ syndrome, with variable eosinophil infiltration. In contrast, systemic involvement or organ damage is untypical for Wells’ syndrome, but this is one important characteristic in the definition for HES. Clinical and histopathological findings of both syndromes may overlap up to the fact that cutaneous manifestations of eosinophilic cellulitis can be part of HES (19–21).
Therapy normally relies on topical or systemic corticosteroids, dapsone or systemic antihistamines (2, 16, 22). In our case, the combination of systemic corticosteroids and dapsone seemed to be effective. It has been described that the anti-oxidative additives of dapsone can suppress free radicals in eosinophils (18). If a causative agent can be identified, it may be useful to treat the underlying condition (7, 10). In our case, chemotherapy of the AILD and afterwards autologous stem cell transplantation led to regression of the skin lesions.
Other therapeutic options include antimicrobial agents (11, 16), colchicines (23), interferon-α (24), antimalarial drugs or immunosuppressive agents, such as cyclosporine or azathioprine (3, 25, 26). New therapeutic options may be tyrosine kinase inhibitors, such as imatinib. This treatment has been shown significantly to decrease blood eosinophil cell counts in patients who carry the FIP1L1-PDGFRA fusion gene. This therapy was successfully carried out in a patient with eosinophilic cellulitis in association with chronic eosinophilic leukaemia (27).
In conclusion, depending on the clinical findings, Wells’ syndrome is an important differential diagnosis to erysipelas, urticaria or drug eruption, and should be considered in particular in patients with neoplasms and/or elevated cell counts of eosinophilic cells in the blood or bone marrow. It is probable that Wells’ syndrome occurs more frequently than is currently assumed.
Conflict of interest: None to declare.
REFERENCES
1. Wells GC, Smith NP. Eosinophilic cellulitis. Br J Dermatol 1979; 100: 101–109.
2. Caputo R, Marzano AV, Vezzoli P, Lunardon L. Wells Syndrome in adults and children. Arch Dermatol 2006; 142: 1157–1161.
3. Moossavi M, Mehregan DR. Wells’ syndrome: a clinical and histopathologic review of seven cases. Int J Dermatol 2003; 42: 62–67.
4. Steffen C. Gorge Crichton Wells – eosinophilic cellulitis (Wells syndrome). Am J Dermatopathol 2002; 24: 164–165.
5. Calvert J, Shors AR, Hronung RL, Poorsattar SP, Sidbury R. Relapse of Wells’ syndrome in a child after tetanus-diphteria immunization. J Am Acad Dermatol 2006; 54: 232–233.
6. Brunner M, Göring HD. Eosinophile Zellulitis (Wells’ Syndrom). Akt Dermatol 2003; 29: 243–246.
7. Hirsch K, Ludwig RJ, Wolter M, Zollner TM, Hardt K, Kaufmann R, Boehncke WH. Eosinophilic cellulitis (Wells’ syndrome) associated with colon carcinoma. JDDG 2005; 3: 530–531.
8. Boura P, Sarantopoulos A, Lefaki I, Skendros P, Papadopoulos P. Eosinophilic cellulitis (Wells’ syndrome) as a cutaneous reaction to the administration of adalimumab. Ann Rheum Dis 2006; 65: 839–840.
9. Winfield H, Lain E, Horn T, Hoskyn J. Eosinophilic cellulitis-like reaction to subcutaneous etanercept injection. Arch Dermatol 2006; 142: 218–220.
10. Zeeli T, Feinmesser M, Segal R, David M. Insect-bite-like Wells’ syndrome in association with mantle-zone lymphoma. Br J Dermatol 2006; 155: 614–616.
11. Gilliam AE, Bruckner AL, Howard RM, Lee BP, Wu S, Frieden IJ. Bullous “cellulitis” with eosinophilia: case report and review of Wells’ syndrome in childhood. Pediatrics 2005; 116: 149–155.
12. Mould AW, Matthaei KI, Young IG, Foster PS. Relationship between interleukin-5 and eotaxin in regulating blood and tissue eosinophilia in mice. J Clin Invest 1997; 99: 1064–1071.
13. Trueb RM, Lubbe J, Torricelli R, Panizzon RG, Wuthrich B, Burg G. Eosinophilic myositis with eosinophilic cellulitis-like skin lesions. Association with increased serum levels of eosinophil cationic protein and interleukin-5. Arch Dermatol 1997; 133: 225–227.
14. España A, Sanz ML, Sola J, Gil P. Wells’ Syndrome (eosinophilic cellulites): correlation between clinical activity, eosinophil levels, eosinophil cation protein and interleukin-5. Br J Dermatol 1999; 140: 127–130.
15. Peters MS, Schroeter AL, Gleich GJ. Immunofluorescence identification of eosinophil granule major basic protein in the flame figures of Wells‘ syndrome. Br J Dermatol 1983; 109: 141–148.
16. Aberer W, Konrad K, Wolff K. Wells‘ syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol 1988; 18: 105–114.
17. Brehmer-Andersson E, Kaaman T, Skog E, Frithz A. The histopathogenesis of the flame figure in Wells‘ syndrome based on five cases. Acta Derm Venereol 1986; 66: 213–219.
18. Plötz SG, Abeck D, Behrendt H, Simon HU, Ring J. Eosinophile Zellulitis (Wells-Syndrom). Hautarzt 2000; 51: 182–186.
19. Tsuji Y, Kawashima T, Yokota K, Tateishi Y, Kobayashi H, Itoh A, Shimizu H. Wells‘ syndrome as a manifestation of hypereosinophilic syndrome. Br J Dermatol 2002; 147: 808–840.
20. Bogenrieder T, Griese DP, Schiffner R, Buttner R, Riegger GA, Hohenleutner U, Landthaler M. Wells‘ syndrome associated with idiopathic hypereosinophilic syndrome. Br J Dermatol 1997; 137: 978–982.
21. Fujii K, Tanabe H, Kanno Y, Konishi K, Ohgou N. Eosinophilic cellulitis as a cutaneous manifestation of idiopathic hypereosinophilic syndrome. J Am Acad Dermatol 2003; 49: 1174–1177
22. Odia SG, Puerschel W, Worret WI, Rakoski J. Hypereosinophilic cellulitis (Wells‘ syndrome) resembling urticaria. Acta Dermatovenerologica Alpina Panomica et Adriatica (APA) 1994; 4: 193–196.
23. Pachet P, Laso-Dosal F, de la Brassine M. Wells‘ syndrome: report of 2 cases. Dermatology, 1992; 184: 139–141.
24. Husak R, Goerdt S, Orfanos CE. Interferon α treatment of a patient with eosinophilic cellulitis and HIV infection. N Eng J Med 1997; 337: 641–642.
25. Herr H, Jai-Kyoung K. Eosinophilic cellulitis (Wells‘ syndrome) successfully treated with low-dose cyclosporine. J Korean Med Sci 2001; 16: 664–668.
26. Karabudak O, Dogan B, Taskapan O, Harmanyeri Y. Eosinophilic cellulitis presented with semicircular pattern. J Dermatol 2006; 33: 798–801.
27. Davis RF, Dusanjh P, Majid A, Fletcher A, Wardlaw A, Siebert R, et al. Eosinophilic cellulitis as a presenting feature of chronic eosinophilic leukaemia, secondary to a deletion on chromosome 4q12 creating the FIP1L1-PDGFRA fusion gene. Br J Dermatol 2006; 154: 1074–1108.