Jae Eun Choi, Hyo Hyun Ahn, Young Chul Kye and Soo Nam Kim*
Department of Dermatology, Korea University Anam Hospital, #126-1, 5-Ka, Anam-dong, Sungbuk-ku, Seoul, 136-705, Korea. *E-mail: grace79@korea.ac.kr
Accepted September 13, 2007.



Jae Eun Choi, Hyo Hyun Ahn, Young Chul Kye and Soo Nam Kim*
Department of Dermatology, Korea University Anam Hospital, #126-1, 5-Ka, Anam-dong, Sungbuk-ku, Seoul, 136-705, Korea. *E-mail: grace79@korea.ac.kr
Accepted September 13, 2007.
Sir,
Precursor B-cell lymphoblastic lymphoma (B-LBL) is a rare subtype of lymphoblastic lymphoma (LBL). LBL is the most aggressive, high-grade subtype of non-Hodgkin’s lymphoma (NHL), accounting for only 5% of all NHL. Most cases of LBL are of T-cell lineage, and B-LBL comprises no more than 10% of LBL. B-LBL is composed of immature lymphocytes that demonstrate lymphoblastic morphology and express precursor and B-cell markers. Unlike precursor T-cell lymphoblastic lymphoma (T-LBL), relatively little is known about B-LBL and only a few cases have been reported in the dermatological literature (1–7). Clinically, B-LBL usually involves the skin, lymph nodes and bone, and occurs in childhood or young adulthood (1). Korea has a much lower rate of B-cell lymphoma than Western countries (3). We describe here a rare case of B-LBL.
CASE REPORT
An 8-year-old boy presented with a slowly growing solitary plaque on his left mandibular area, which had been present for 6 months. His medical history was otherwise unremarkable and he was not on any medication. He had no fever, weight loss or other symptoms.
Physical examination revealed a dusky erythematous, firm, fixed, infiltrative tender plaque, measuring 5.5 × 5.0 cm on his left mandibular area (Fig. 1). There were no palpable lymph nodes or hepatosplenomegaly. Biopsy showed a diffuse dermal infiltration of uniform, small to medium-sized round cells, which were mitotically active lymphoid cells with irregular nuclei, condensed chromatin, inconspicuous nucleoli and scant cytoplasm. The epidermis was spared and separated from the neoplastic cells by a thin layer of unaffected dermis (Fig. 2).
Fig. 1. Solitary, firm, fixed, infiltrative plaque on the left mandibular area.
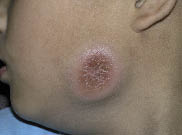
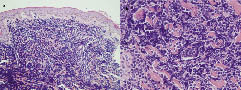
Fig. 2. (a) Diffuse dermal infiltration of uniform, small to medium-sized lymphoid cells. Epidermis is spared and separated from the region by a Grenz zone. (b) The lymphoid cells have condensed chromatin, inconspicuous nucleoli with scant cytoplasm and a high mitotic rate.
Immunostaining demonstrated that most of the lymphocytes stained positively for TdT, CD79a and CD10, and negatively for CD3, CD20 and myeloperoxidase. Immunostaining for Ki-67 showed positive immunostaining of more than 95% of the lymphocytes (Fig. 3).
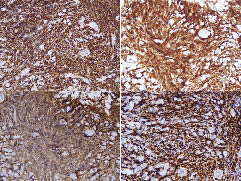
Fig. 3. The neoplastic cells stain with (a) TdT, (b) CD79a and (c) CD10, but not stained with CD3, CD20 or myeloperoxidase. (d) Immunostaining with Ki-67 shows positive immunostaining for more than 95% of the lymphocytes.
A complete blood cell count showed a haemoglobin level of 13.3 g/dl, a leukocyte count of 7870 /mm3 and platelet count of 260000 /mm3. The serum level of lactate dehydrogenase (LDH) was raised, at 462 IU/l. Computerized tomography (CT) scans showed multiple enlarged lymph nodes on the mesenteric root and a few enlarged regional lymph nodes. Bone marrow biopsy showed overall 20–30% of marrow cellularity with no involvement of malignant lymphoma. Cerebrospinal fluid cytology did not reveal any involvement.
Treatment followed a Berlin-Frankfurt-Münster (BFM)-NHL protocol combination chemotherapy for localized LBL (8). Within 9 weeks of induction, protocol I (was given prednisone, vincristine, daunorubicin, asparaginase, cyclophosphamide, cytarabine, mercaptopurine and intrathecal methotrexate), the involved skin appeared to be in remission, and the patient was then given consolidation protocol M (mercaptopurine, methotrexate and intrathecal methotrexate) for 8 weeks. Now, 13 months after the initial diagnosis, the patient has been receiving maintenance therapy (daily mercaptopurine and weekly methotrexate) and most of the involved lymph nodes have disappeared or reduced in size, as assessed by CT scan, indicating a good response to the chemotherapy.
DISCUSSION
LBL, like acute lymphoblastic leukaemia (ALL), comprises high-grade neoplasms arising from precursor lymphocytes of B-cell or T-cell lineage. The neoplastic cells of LBL are morphologically indistinguishable from those of ALL and they are grouped together in the WHO classification. Commonly used criteria to discriminate LBL from ALL are focal (< 25%) or absent bone marrow involvement and absence of peripheral blood involvement (1). According to the Revised European-American Classification of Lymphoid Neoplasms, ALL and LBL account for 80% and 20% of neoplastic proliferation of lymphoblasts, respectively, and less than 10% of cases of LBL are of B-cell lineage, unlike ALL in which 85% express B-cell markers (1, 9). In Korea the incidence of B-cell lymphoma (3%) is very low compared with Western countries, while the proportion of T-cell subtypes is much higher (96%) than in Europe (79.2–81.1%) (10).
B-LBL seems to be a disease of young people, with a preponderance of young females, in contrast to T-LBL (7). Skin, lymph node and bone involvement are observed most frequently in B-LBL, whereas bone marrow, CNS and mediastinal involvement are more common in T-LBL. The cutaneous manifestations are usually red to purple coloured nodules or papules in the head and neck area. Most patients seem to have no constitutional symptoms (1–5).
B-LBL morphologically belongs to a category of tumours known as small round cell tumours (7). It shows diffuse or nodal dermal homogeneous infiltration of the small to medium-sized lymphoid cells in the dermis and subcutis. They are composed of mitotically active immature lymphoid cells with fine chromatin, inconspicuous or small nucleoli, and scant basophilic cytoplasm. A focal or diffuse starry sky pattern, necrotic foci or karyorrhectic debris may be seen. A narrow Grenz zone separates the tumour from the overlying normal epidermis (1, 2, 11). In children, this category of small round cell tumours includes Ewing’s sarcoma/primitive neuroectodermal tumour (ES/PNET), rhabdomyosarcoma, neuroblastoma, Wilms’ tumour, granulocytic sarcoma and malignant lymphoma (7). As treatment modalities are completely different for each tumour, the exact differential diagnosis is important, which is largely dependent on immunophenotyping.
Immunophenotypically, B-LBL demonstrates lymphoblastic morphology, with expression of TdT, CD99, HLA-DR and a tendency to express B-cell markers, such as CD79a, CD19, CD20, CD22 and CD10, but is negative for T-cell antigens (12). Among the B-cell antigens, CD19, CD79a, CD10 are more commonly expressed and TdT is expressed in more than 90% of cases, and these are reliable confirming markers for B-LBL. CD20, CD22 and CD99 show rather variable reactivity (1). Recent studies have reported that CD179a and CD179b may be important markers for the immunophenotypical diagnosis of B-LBL (10). Our case showed typical clinicopathological and immunophenotypical features of B-LBL.
The distinction between LBL and ES/PNET is of particular importance. This is complicated by an overlap in the immunoperoxidase staining pattern of CD99 positivity. Although it is a sensitive marker for ES/PNET, the majority of LBL also appear to be positive for this marker. However, lymphoid markers such as TdT, CD43, or CD79a, which are negative in ES/PNET and positive in B-LBL, distinguish them (7). Cutaneous rhabdomyosarcoma is differentiated by positive muscle markers and negative lymphoid markers and cutaneous neuroblastoma usually by negative lymphoid markers. B-LBL is differentiated from T-LBL by its reactivity for at least one pan-T-cell marker (CD2, CD3, CD5 or CD7). The blastoid variant of mantle cell lymphoma is histologically identical in appearance to LBL, but it does not express the immature markers such as TdT and CD34 and clinically it affects older people in their sixth or seventh decades, with a strong male predominance (13, 14).
B-LBL, which naturally has a poor prognosis, is potentially curable with current multi-agent systemic chemotherapy. Ineffective treatment appears to be an important cause of recurrence and poor prognosis (1, 2, 15, 16). Correct diagnosis, including histopathology, immunophenotyping of complete B-cell and T-cell profiles and genetics to differentiate patients with B-LBL from other subtypes of lymphoma, is therefore essential in order to provide appropriate treatment and the chance of a better prognosis. Our patient was treated with BFM-NHL protocol combination chemotherapy with a favourable response. This has proven efficacious for localized B-LBL, with a 5-year event-free survival of approximately 85% (8).
REFERENCES
1. Maitra A, McKenna RW, Weinberg AG, Schneider NR, Kroft SH. Precursor B-cell lymphoblastic lymphoma. A study of nine cases lacking blood and bone marrow involvement and review of the literature. Am J Clin Pathol 2001; 115: 868.
2. Lin P, Jones D, Dorfman DM, Medeiros LJ. Precursor B-cell lymphoblastic lymphoma: a predominantly extranodal tumor with low propensity for leukemic involvement. Am J Surg Pathol 2000; 24: 1480.
3. Kim J-Y, Kim YC, Lee E-S. Precursor B-cell lymphoblastic lymphoma involving the skin. J Cutan Pathol 2006; 33: 649–653.
4. Zanation AM, Ebert CS Jr, Coffey CS, Dubin MG, Rose AS. Precursor B-cell lymphoblastic lymphoma presenting as an isolated external ear swelling in a two-year-old child. Int J Pediatr Otorhinolaryngo 2005; 69: 695–699.
5. Recine M, Castellano-Sanchez AA, Sheldon J, Schwartz M, Cabello-Inchausti B. Precursor B-cell lymphoblastic lymphoma/leukemia presenting as osteoblastic bone lesions. Ann Diagn Pathol 2002; 6: 236–243.
6. Shiozawa Y, Kiyokawa N, Fujimura J, Suzuki K, Yarita Y, Fujimoto J, et al. Primary malignant lymphoma of the central nervous system in an immunocompetent child: a case report. J Pediatr Hematol Oncol 2005; 27: 561–564.
7. Kahwash SB, Qualman SJ. Cutaneous lymphoblastic lymphoma in children: report of six cases with precursor B-cell lineage. Pediatr Dev Pathol 2002; 5: 45–53.
8. Atlas M. Non-hodgkin lymphoma. In: Lanzkowsky P, editor. Manual of pediatric hematology and oncology. London: Elsevier Academic Press, 2005: p. 500–502.
9. Kiyokawa N, Sekino T, Matsui T, Takenouchi H, Mimori K, Tang WR, et al. Diagnostic importance of CD179a/B markers of precursor B-cell lymphoblastic lymphoma. Mod Pathol 2004; 17: 423.
10. Lee MW and the Korean Dermatopathology Research Group. Characteristics of cutaneous lymphomas in Korea. Clin Exp Dermatol 2003; 28: 639–646.
11. Sander CA, Medeiros LJ, Abruzzo LV, Horak ID, Jaffe ES. Lymphoblastic lymphoma presenting in cutaneous sites: a clinicopathologic analysis of six cases. J Am Acad Dermatol 1991; 25: 1023.
12. Soslow RA, Baergen RN, Warnke RA. B-lineage lymphoblastic lymphoma is a clinicopathologic entity distinct from other histologically similar aggressive lymphomas with blastic morphology. Cancer 1999; 85: 2648.
13. Soslow RA, Zukerberg LR, Harris NL, Warnke RA. Bcl-1 (PRAD- 1/Cyclin D-1) overexpression distinguishes blastoid variant of mantle cell lymphoma from B-lineage lymphoblastic lymphoma. Mod Pathol 1997; 10: 810–817.
14. Soslow RA, Bhargava V, Warnke RA. MIC2, TdT, bcl-2 and CD34 expression in paraffin-embedded high grade lymphoma/acute lymphoblastic leukemia distinguishes between distinct clinicopathologic entities. Hum Pathol 1997; 28: 1158–1165.
15. Picozzi VJ. Lymphoblastic lymphoma. Semin Oncol 1990; 17: 96.
16. Neth O, Seidemann K, Jansen P, Mann G, Tiemann M, Ludwig WD, et al. Precursor B-cell lymphoblastic lymphoma in childhood and adolescence: clinical features, treatment, and results in trials NHL-BFM 86 and 90. Med Pediatr Oncol 2000; 35: 20–27.

