Christiane Schlegel1, Gisela Metzler1, Walter Burgdorf2 and Martin Schaller1
Department of Dermatology, 1Eberhard Karls University, Tübingen, and 2Ludwig-Maximilians-University Munich, Munich, Germany



Christiane Schlegel1, Gisela Metzler1, Walter Burgdorf2 and Martin Schaller1
Department of Dermatology, 1Eberhard Karls University, Tübingen, and 2Ludwig-Maximilians-University Munich, Munich, Germany
Progressive mucinous histiocytosis is a very rare, benign, non-Langerhans’ cell histiocytosis limited to the skin. In total ten patients (all women) in four families and three sporadic cases have been reported. We report here the first published case of a male patient with progressive mucinous histiocytosis. The multiple red papules on the scalp and forearms were asymptomatic and had slowly increased over approximately the past 20 years. The patient’s mother had similar lesions. Histological examination revealed nodules in the dermis with histiocytes and mucin deposition. The histiocytes stained positively with CD31 and negative with CD34, CAM 5.2, PGM-1 and factor XIIIa. Ultrastructurally, the histiocytes showed numerous circular myelin bodies and zebra bodies reminiscent of those seen in lysosomal storage diseases. The genetic transmission of hereditary progressive mucinous histiocytosis remains unclear; we assume an autosomal dominant transmission with some hormonal factor that makes hereditary progressive mucinous histiocytosis more likely in women. Key words: familial occurrence; hereditary progressive mucinous histiocytosis; mucin.
(Accepted August 21, 2009.)
Acta Derm Venereol 2010; 90: 65–67.
Martin Schaller, Department of Dermatology Tübingen, Eberhard Karls University, Liebermeisterstr. 25, DE-72076 Tübingen, Germany. E-mail: martin.schaller@med.uni-tuebingen.de
The histiocytoses are a heterogeneous group of diseases divided by the Histiocyte Society into class I (Langerhans’ cell histiocytoses), class II (histiocytoses of mononuclear phagocytes other than Langerhans’ cells) and class III (all the malignant histiocytoses, derived from Langerhans’ or non-Langerhans’ cells). Class II encompasses a number of rare and often poorly understood disorders, including hereditary progressive mucinous histiocytosis (HPMH). Our patients’ clinical, histological and ultrastructural features have been described previously as HPMH, a rare often hereditary disorder characterized by multiple persistent papules with prominent mucinosis (1, 2), which has been only reported in ten female patients.
We report here two additional patients with clinical, histological and ultrastructural features consistent with HPMH, including the first report of a male patient with this disease.
CASE REPORT
A 65-year-old woman and her 40-year-old son presented with multiple small red papules on the extensor aspect of the forearms and the scalp. The son’s papules were completely asymptomatic, but the mother reported marked pruritus on the scalp. The face, trunk and lower extremities were not affected. The mother had first noticed the papules on the forearms approximately 40 years prior to presentation; the son had developed lesions more than 20 years ago. The mother had hypertension, hyperlipidaemia and depression. Her medications were irbesartan, hydrochlorothiazide, acetylsalicylamide, pravastatin and mirtazapine. Her son had no medical problems. Neither patient had systemic signs nor symptoms, such as polydipsia, polyuria or seizures.
On examination, the cutaneous lesions were small red papules, mostly less than 5 mm in diameter (Fig. 1). They were not ulcerated. No mucous membrane lesions were found. Five papules from the mother and two from the son were biopsied; all showed similar histological features. There were well-circumscribed nodules in the upper dermis composed of monomorphous mononuclear cells embedded in a myxoid stroma (Fig. 2). The cells within the nodules were macrophages, which stained positively with CD31 and negatively with CD34, CAM 5.2, PGM-1 and Factor-XIII-a. A few of the cells expressed KP-1. At the ultrastructural level the macrophages showed numerous circular myelin bodies and zebra bodies reminiscent of those seen in lysosomal storage diseases (Fig. 3). Birbeck granules were not seen.

Fig. 1. Numerous erythematous papules on (a) the forearm and (b) the scalp.
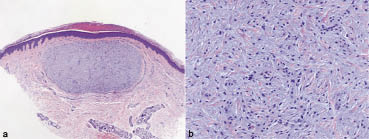
Fig. 2. (a) Well-circumscribed nodule consisting of (b) monomorphous mononuclear cells embedded in a myxoid stroma. Haematoxylin and eosin staining; original magnification × 20 and × 200, respectively.
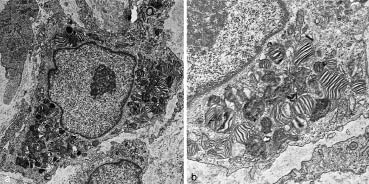
Fig. 3. Transmission electron microscopy showing (a) a macrophage with (b) numerous circular myelin bodies and zebra bodies. Original magnification × 2000 and × 5000, respectively.
DISCUSSION
On clinical, histological and ultrastructural grounds, our patients most closely resemble HPMH. This peculiar disorder was first described by Bork & Hoede 1988 in an elderly woman, her sister and her niece, who developed papules on the extremities, trunk, face and scalp, usually starting at around 10 years of age (3). The number of papules slowly increased over the years. Extensive laboratory investigation revealed neither paraproteinaemia nor abnormalities of lipid metabolism. Bork subsequently reported a further two patients; a mother and daughter, with lesions that were histologically and ultrastructurally very similar to the original cases. Two further patients, a mother-daughter pair from Germany, began to develop skin-coloured to red-brown asymptomatic papules on the nose, dorsum of the hands, forearms and thighs, starting at about 20 years of age, with no tendency to resolve spontaneously (4).
Two affected women in a family from Spain were a 56-year-old mother and her 19-year-old daughter. They began developing multiple papules and nodules from the age of 11 and 12 years (5). The youngest patient reported with HPMH was a 6-year-old Japanese girl, who had several nodules at birth (6). More recently, Pfister et al. (7) observed the first recorded French HPMH patient, a 48-year-old woman with asymptomatic papules on the dorsum of the hands starting at 11 years of age. Sass et al. (1) observed a sporadic case of HPMH in a 64-year-old woman who had multiple dark-red dome-shaped papules and nodules located mainly on the back of her hands, forearms and thighs. Two additional sporadic cases of adult-onset HPMH were described by Young et al. (8): two unrelated African-American woman, aged 48 and 55 years developed red-brown and flesh-coloured, asymptomatic papules on the face, the arms and the legs without truncal, mucosal or visceral involvement. Antoni-Bach et al. (9) reported another family with HPMH. A 49-year-old woman presented with asymptomatic papules on the dorsum of her hands known since childhood. Her mother and two sisters had the same condition, while her two sons, her brother and her nephews did not.
Histological skin examinations of all these patients showed features identical to our case: well-circumscribed non-encapsulated aggregates of oval and spindle-shaped macrophages and mucin in the upper and middle dermis. Immunohistochemical staining for S100, Factor XIIIa, MAC-387 and LN 5 was negative. In the literature immunohistochemical studies on formalin-fixed paraffin-embedded tissue sections (Table I) generally showed positive labelling of the cells with alpha-1 antitrypsin, and factor XIIIa, along with negative staining for S100 protein, CD1a and CD34 (1). Results for the macrophage-related antigen CD68 (KP1) have been variable. Bork (5) reported negative staining, whereas Schroder et al. (4) stated that the macrophages showed strong or moderate labelling. In our patients the histiocytes were negative for CD68. Correspondingly, in all published cases, stainings with the Langerhans’ cell markers CD1a, S100 and CD34 were negative.
Table I. Staining pattern of the histiocytes in hereditary progressive mucinous histiocytosis
| Our patient | Previous patients (ref) |
| CD68 negative | CD68 negative (5) or positive (4) |
| CD1a negative | CD1a negative (1) |
| S100 negative | S100 negative (1) |
| Factor XIIIa negative | Factor XIIIa negative (9) |
The main ultrastructural features common to the lesions previously described, and those in our patient, were multiple cytoplasmic inclusions in the histiocytes, appearing as zebra bodies and closely resembling a lysosomal storage disease. The aetiology of these structures remains unknown.
The main differential diagnoses of HPMH include acral papular mucinosis, scleromyxoedema, granuloma annulare, dermatofibromas (especially mucinous dermatofibromas), multicentric reticulohistiocytosis, eruptive histiocytoma and other forms of non-Langerhans’ histiocytosis. The relationship between the various histiocytic disorders currently classified under class II histiocytoses remains unclear. HPMH may represent a mucinous variant of eruptive histiocytoma, but in contrast to HPMH, eruptive histiocytomas usually undergo spontaneous resolution, and there is a lack of familial history. Antal et al. (10) described a 20-year-old woman with multiple eruptive myxoid dermatofibromas. She presented with multiple papules and nodules that had developed over a period of 9 years (10). Skin biopsies showed plump to spindle-shaped fibrocytes between thickened bundles of collagen and an excessive accumulation of mucin within the dermis. Thirty percent of the lesion represented by spindle cells were positive for vimentin; CD34 showed regular staining of endothelial cells; CD1a, S100 protein, CD68 and SMA were negative.
The genetic transmission of HPMH remains unclear. X-linked recessive transmission of HPMH can be ruled out, since if this was the case, women would never be affected. Genetic diseases with X-linked dominant transmission have fewer sons than daughters in larger pedigrees, and the disease is usually more severe in men than women. The male patient described here, however, was not dramatically worse than his mother. We therefore assume an autosomal dominant transmission with some hormonal factor that makes HPMH more likely in women.
REFERENCES

