Wolfgang Bauer1, Daniel Spazierer1, Irene Klein1, Georg Stary1, Stefan Wöhrl1, Stephan N. Wagner1, Robert Knobler2, Leonhard Müllauer3 and Georg Stingl1
Department for Dermatology, 1Division of Immunology, Allergy and Infectious Diseases, 2Division of General Dermatology and 3Department for Pathology, Medical University of Vienna/General Hospital, Waehringerguertel 18–20, AT-1090 Vienna, Austria. E-mail: wolfgang.bauer@meduniwien.ac.at
Accepted May 9, 2011.
Cutaneous T-cell lymphomas (CTCLs) are a heterogeneous group of lymphoproliferative disorders of the skin (1). Sézary syndrome is a rare variant with an aggressive clinical behaviour, presenting with erythroderma, generalized lymphadenopathy and leukaemic blood involvement. The neoplastic T cells are considered to be CD4+ TCRαβ+ memory cells with skin-homing properties and a Th2 phenotype.
Other forms of CTCL frequently show expression of a cytotoxic phenotype (e.g. primary cutaneous CD30+ lymphoproliferative disorders, subcutaneous panniculitis-like lymphoma, extranodal NK-/T-cell lymphoma). Among them, primary cutaneous TCRγδ+ T-cell lymphomas (PCGD-TCL) encompass a rare new entity in the current WHO-EORTC classification (1). They are characterized clinically by disseminated plaques, nodules or tumours with a poor prognosis.
We describe here a case of a peripheral TCRγδ+ T-cell lymphoma with a Th2 phenotype and a clinical manifestation resembling Sézary syndrome.
CASE REPORT
A 78-year-old Caucasian man was admitted to our hospital with a 5-month history of recurrent recalcitrant pruritus and erythroderma that had been interpreted previously as a drug hypersensitivity reaction or as late-onset atopic dermatitis. The patient had been treated repeatedly with local and systemic corticosteroids as well as antihistamines, but had relapsed invariably after tapering of treatment. On admission the patient presented with a generalized erythema, with lichenification of flexural areas, minimal scaling and excoriations, but exhibited neither hair or nail abnormalities nor palmoplantar hyperkeratosis. Inguinal lymph nodes on the right side were palpable, indolent and freely movable. They were described as reactively enlarged (4 × 1.5 cm) in an ultrasonic examination. Primary laboratory findings revealed lymphopaenia with a slight CD4/CD8 T-cell imbalance (4/1), highly elevated IgE levels (532 kU/l) and eosinophilia (6% of leukocytes). Histology of a skin biopsy showed features consistent with eczema. The past medical history of the patient included a myocardial infarction and an allergic rhinoconcunjctivitis. Clinical differential diagnoses included a drug hypersensitivity reaction, generalized atopic dermatitis, as well as Sézary syndrome and erythrodermic mycosis fungoides.
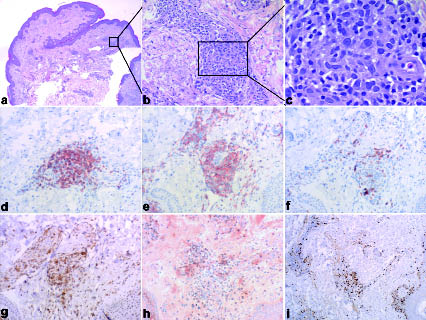
Within one month after stopping treatment with local or systemic glucocorticoids for diagnostic purposes the disease exacerbated, with generalized lymphadenopathy, flaming red erythroderma, insomnia due to pruritus, hypereosinophilia (up to 63% of leucocytes) and a shift of the CD4/CD8 T-cell ratio in peripheral blood up to 12. Sixty percent of lymphocytes (877 cells/µl) stained positive for CD3, CD4 and CD5, with loss of expression of the pan-T-cell antigens CD2 and CD7. Surprisingly and in contrast to Sézary syndrome, the malignant cells did not display a cerebriform nucleus and expressed the γδTCR. No B cells were detectable in the patient’s blood. Diagnostic imaging using computed tomography scans and positron emission tomography confirmed pathological enlargement of cervical, thoracic and abdominal lymph nodes without involvement of any other organs. Despite significant clinical involvement, skin pathology (Fig. 1 a–c) showed only a sparse perivascular dermal infiltrate of atypical, proliferating cells staining positive for CD3 and CD4 but not for CD7, CD8 and βF1 (Fig. 1 d–i). Epidermotropism or involvement of the subcutaneous tissue could not be seen. In contrast, histopathological examination of a lymph node showed complete destruction of its architecture by a dense interfollicular infiltrate of atypical lymphoid cells (Fig. 2) expressing the same marker profile as the skin-infiltrating cells (data not shown). A bone marrow biopsy of the iliacal crest showed discrete infiltration with the same atypical lymphocytes and an otherwise normal haematopoiesis. Identical clonal TCRγ gene rearrangement was detected in blood, lymph node and bone marrow, but not in skin, by heteroduplex PCR analysis. Cytodiagnostics revealed a complex aberrant phenotype. CHIC2 deletion (FISH) or FIP1L1-PDGFRα fusion (PCR) was ruled out as a cause for eosinophilia. Serologically there was no association with HTLV-1 or Epstein-Barr virus infection. The disease was classified as a peripheral T-cell lymphoma, unspecified according to the WHO-EORTC classification (1).
Fig. 1. (a) Histopathology of a skin specimen from the right elbow flexure demonstrating a sparse perivascular infiltrate in the dermis without any epidermotropism. (b, c) The atypical lymphoid cells are medium in size with enlarged irregular nuclei (haematoxylin-eosin, original magnification (a) ×20, (b) ×200, (c) ×600). Immunohistochemical analysis of formalin-fixed, paraffin-embedded sections showed immunoreactivity of the infiltrating cells with antibodies against (d) CD3 and (e) CD4, but not with (f) CD8, (g) CD7 and (h) βF1. Proliferation is depicted by staining with antibody Mib1 (i) (original magnification ×200, i: ×100).
Fig. 2. (a) Lymph node histopathology (right inguinal lymph node) reveals a destructed architecture with a dense interfollicular infiltrate of atypical lymphoid cells that penetrates the capsule. (b) The lymphoid cells were medium in size with enlarged nuclei and in parts with several nucleoli (haematoyxlin-eosin, original magnification (a) ×10, (b) ×600).
Further phenotypic characterization of the patient’s peripheral blood mononuclear cells (PBMC) was performed by immunostaining and fluorescence-activated cell sorting (FACS) analysis. The malignant clone was shown to mainly express CD45RA, but, to a lesser extent, also CD45RO, the TCR Vδ1 chain, CCR4 and CCR7. The cells lacked any expression of cytotoxic molecules (granzyme B, perforin), of NKG2D, CD56, CD57, Foxp3 or GITR (data not shown). Stimulation of the patient’s PBMC with anti-CD3 and anti-CD28 monoclonal antibodies led to higher production of IL-4 and IL-13 with virtually absent IFN-γ production compared with PBMC of healthy volunteers (Fig. 3A). As a result, massive eosinophilia was demonstrable and correlated with the absolute numbers of malignant T cells in the patient’s blood during therapy (data not shown).
To verify cytokine production by the tumour cells themselves and rule out influences of other mononuclear cells, this was confirmed by quantitative real-time PCR (qPCR) of freshly isolated and FACS-sorted malignant CD3+CD4+TCRγδ+CD7– T cells (Fig. 3B). In addition, culture of sorted lymphoma cells was successful in the presence of IL-7. They could be propagated for more than 25 passages with minimal change of phenotype, most notably gaining expression of CLA by all cells and loss of CD4 by 50% of cells (data not shown). qPCR after in vitro stimulation confirmed the sustained expression of Th2 cytokines (Fig. 3C). Titration of cultivated tumour cells to PBMC of healthy donors and stimulation with poke-weed mitogen (PWM) led to increased differentiation of B cells to plasma cells (Fig. 3D), further corroborating their Th2 function and providing a possible explanation for the observed absence of peripheral blood B cells.
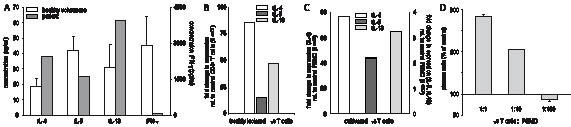
Fig. 3. The malignant γδ T cells have a Th2 phenotype. (A) Peripheral blood mononuclear cells (PBMC) of the patient produce higher amounts of Th2 cytokines than those of healthy volunteers and do not produce significant amounts of interferon (IFN)-γ. PBMC of the patient or of healthy volunteers were stimulated with anti-CD3 (1µg/ml) and anti-CD28 (0.5 µg/ml) mAbs for 40 h. Cytokine production was assessed by enzyme-linked immunoassay (ELISA) of cell culture supernatants. (B) Freshly isolated malignant γδ T cells produce high amounts of prototypic Th2 cytokines IL-4, IL-5 and IL-13. Expression of cytokine mRNAs indicated was measured by qPCR of sorted malignant γδ T cells. Data are normalized to β2-microglobulin, and represent the fold changes relative to sorted CD3+CD4+ T cells from a healthy volunteer. (C) Cultivated malignant γδ T cells maintain their Th2 phenotype. After propagation in vitro for > 15 passages, γδ T cells were stimulated with anti-CD3 and anti-CD28 mAbs and cytokine expression was measured by qPCR. Data represent the fold changes relative to PBMC from a healthy volunteer stimulated in the same way. (D) Malignant γδ T cells provide help for the differentiation of B cells to plasma cells. PBMC from healthy donors were cultivated in the presence of poke-weed mitogen and titrated amounts of cultivated γδ T cells for 10 days. Plasma cell differentiation was measured by staining with mAbs against CD138 and CD38 and fluorescence-activated cell sorting analysis.
Due to systemic involvement and the known aggressive course of γδ T-cell lymphomas, chemotherapy with cyclophosphamide, liposomal doxorubicin, vincristine and prednisolone (CHOP) was initiated. Despite a partial response this treatment had to be stopped after five cycles due to repeated septicaemia. Subsequent treatment with extracorporeal photopheresis in conjunction with ultraviolet B (UVB) radiation and systemic glucocorticosteroids resulted in a stable disease of short duration. The patient died due to disease progression 8 months after initial diagnosis.
Discussion
Malignant proliferations of γδ T cells represent rare cases of T-cell lymphomas. The current WHO classification recognizes two main entities: hepatosplenic and PCGD-TCL, the latter being a newly defined subtype of peripheral T-cell lymphomas, not otherwise specified (PTCL-NOS) (1, 2). T-cell large granular lymphocytic leukaemia, extranodal NK-/T-cell lymphoma and other PTCL-NOS can also exhibit a TCRγδ+ phenotype. PCGD-TCL preferentially manifests on the extremities, usually in the form of plaques, nodules or tumours, corresponding histologically to an infiltration with atypical lymphocytes either in the epidermis, dermis or subcutaneous tissue, with possible co-occurrence of these patterns. The malignant cells display strong expression of cytotoxic molecules (TIA1, granzyme B, perforin) in addition to a CD3+, CD2+, CD5–, CD7+/–, CD56+, CD4–, CD8+/– phenotype.
In contrast to PCGD-TCL or other previously reported γδ T-cell lymphomas the case of a peripheral TCRγδ+ T-cell lymphoma described here displayed a completely different clinical manifestation and phenotype resembling Sézary syndrome. The tumour cells were shown to be CD4+ Th2 cells lacking the expression of cytotoxic molecules, which is extremely unusual for a TCRγδ+ T-cell lymphoma (3, 4). We ultimately cannot rule out the primary manifestation of the lymphoma in the skin and thereby a variant of cutaneous γδ T cell lymphoma. Nevertheless, the clinical manifestation with erythroderma, pruritus, generalized lymphadenopathy and leukaemia matches the classical definition of Sézary syndrome (5). The neoplastic T cells from patients with Sézary syndrome were first described as helper cells in the 1970s (6) and are nowadays considered to be central memory Th2 cells with skin-homing properties (7). Strikingly the tumour cells in our patient reflect this phenotype. The high amounts of Th2 cytokines in blood and skin of patients with Sézary syndrome has been made responsible for the marked immunosuppression observed in this disease (8). On the other hand, regulatory T cells might contribute to this effect, as evidenced by the description of neoplastic T cells expressing the transcription factor FoxP3 (9) and being functionally suppressive in a subset of patients with Sézary syndrome (10). Thus, Sézary syndrome might not even be considered a homogeneous entity, but possibly a common clinical manifestation of transformed regulatory or Th2 cells with skin homing properties and independent of the expressed TCR. Following this, the prognosis of the disease might vary, as depicted by the highly aggressive lymphoma of our patient. This highlights the need for adequate characterization of the tumour cells and development of specific treatment options.
AcknowledgEment
We are indebted to Dr Lorenzo Cerroni for reviewing biopsy specimens.
References