


Division of Dermatology and Venereology, Geneva University Hospitals, rue Gabrielle-Perret-Gentil 4, CH-1205 Genève, Switzerland. E-mail: Caroline.deLorenzi@hcuge.ch
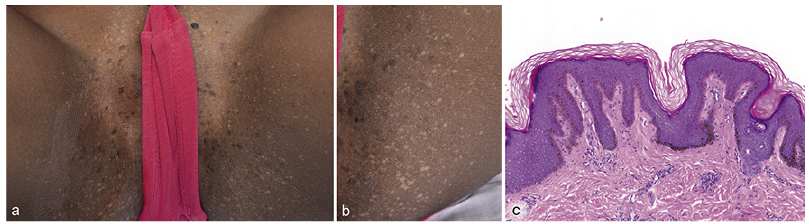
A 7-year-old Eritrean girl presented with a 2-year history of asymptomatic skin lesions that had appeared progressively on her buttocks and genital area. She had no previous personal or familial medical history. Clinical examination revealed hypopigmented macules and small slightly brownish papules on the pubis and inner side of her thighs (Fig. 1 a,b). Initially, an inflammatory papilloma virus (HPV) infection was suspected. A skin biopsy revealed a slight epidermal hyperplasia with pigmentation of the basal cell layer and a focal loss of melanocytes (Fig. 1c). Moderate hyperkeratosis was observed. PCR for HPV was negative.
What is your diagnosis? See next page for answer.
Fig. 1. (a) Hypopigmented macules and brownish papules on the pubis and inner side of the thighs in a 7-year-old girl. (b) Close-up of hypopigmented and brownish papules on the inner side of the thighs. (c) Moderate hyperkeratosis, slight epidermal hyperplasia and focal loss of melanocytes (haematoxylin and eosin (H&E), original magnification ×10).
Acta Derm Venereol 2019; 99: XX
Diagnosis: Dowling-Degos disease
Clinicopathological correlation was consistent with a diagnosis of Dowling-Degos disease (DDD). DDD was first reported as reticular pigmented macules of flexure by Wilson-Jones and Grice in 1974 (1–3). It usually presents as a benign autosomal dominant inherited disease, but sporadic cases have also been reported (1, 2, 4, 5). DDD is more common in females and appears early in adulthood (5).
Classic DDD is characterized by acquired progressive and symmetrical pigmented round-oval macules of light-brown to grey or black colour, which can also be slightly elevated or exhibit slight scaling, occurring in a reticular pattern (5, 6). Rarely, hypopigmented macules and papules have been reported, alone or together with hyperpigmented lesions in the case of generalized DDD (7–9). The skin lesions are restricted to the flexural sites (axillae, groins, submammary folds, neck); generalized cases or cases involving other sites, such as the vulva, have rarely been reported (1, 3, 5, 6). The lesions may be associated with pitted scars (often perioral), hyperkeratotic follicular papules on the neck and upper trunk, and dark comedo-like lesions often seen on the face, neck and upper back (1, 2, 5). Other lesions, such as fingernail dystrophy, epidermoid cysts, keratoacanthomas, pilonidal sinus, seborrhoeic keratosis and hidradenitis suppurativa, have been described (2, 5). There is also an acantholytic variant called Galli-Galli disease (4). Usually, lesions are asymptomatic, but mild pruritus can appear (1, 5).
Diagnosis is established on the clinical-pathological correlation. Histologically, DDD shows filiform elongation of the rete ridges with basal cell pigmentation, but also moderate hyperkeratosis, thinning of the suprapapillary epithelium and dermal melanosis can be observed (5, 6). Hypopigmented lesions show pigment only at the tip of the rete ridges (7, 8).
Because of overlapping clinical features, DDD is believed to be a part of a disease spectrum that includes reticulate acropigmentation of Kitamura, dyschromatosis universalis hereditaria and dyschromatosis symmetrica hereditaria (6, 8).
The pathogenesis of DDD is not well understood, but it is linked to mutations in the KRT5 gene located on chromosome 12, encoding keratin K5 and leading to its loss of function (2, 4–6). Other mutations cause different phenotypic variations, ranging from classic to generalized DDD and Galli-Galli disease (4).
Many therapies have been used without success (topical steroids, hydroquinone, oral retinoids, topical retinoids). No treatments have yet been established to be effective (3, 5, 6).
To conclude, the case reported here is unusual because the patient presented genital lesions of the vulva area with no other clinical characteristics of DDD, and without any family history. No medical or topical causes were found for this condition. The clinicopathological correlation helped us to make the diagnosis. It is important to have good knowledge of the different clinical presentations of DDD, since the disease is rare and challenging, and in order to avoid painful and expensive examinations for the child.


 Click to show fullsize
Click to show fullsize