


Department of Dermatology, Medical Center, University of Freiburg, Faculty of Medicine, University of Freiburg, Hauptstr. 7, DE-79104 Freiburg, Germany. E-mail: franziska.schauer@uniklinik-freiburg.de
Accepted Jun 28, 2019; E-published Jul 8, 2019
Bullous pemphigoid (BP) is a prototypic autoimmune blistering disease, characterized by tissue-bound and circulating autoantibodies directed against collagen XVII/ BP180 and/or dystonin/BP230, both components of the hemidesmosomes involved in dermal–epidermal cohesion (1). Epitope spreading (ES) represents the process of diversification of B-cell and/or T-cell response from the initial dominant epitope to a secondary epitope over time. ES can occur within a single antigen (intramolecular) or involve different antigens (intermolecular) and manifest at various stages of the disease (2, 3). Some ES events appear to be quite common, e.g. shifting between pemphigus vulgaris and pemphigus foliaceus (PF), whereas others are rare. ES, for example, has also been shown in 17 of 35 patients with BP in a multicentre prospective study with a 12-month observation period. The ES occurred preferentially at an early stage of the disease, shaping the course of BP disease (4). However, ES phenomena may also lead to disease transition. We report here a patient with long-standing BP in clinical, but not serological remission, who developed typical PF 13 years after diagnosis of BP. Although a rare event of second autoimmunity cannot be excluded, the serological IgG subclass shift, with transient presentation of features of both BP and PF, is more indicative for epitope spreading.
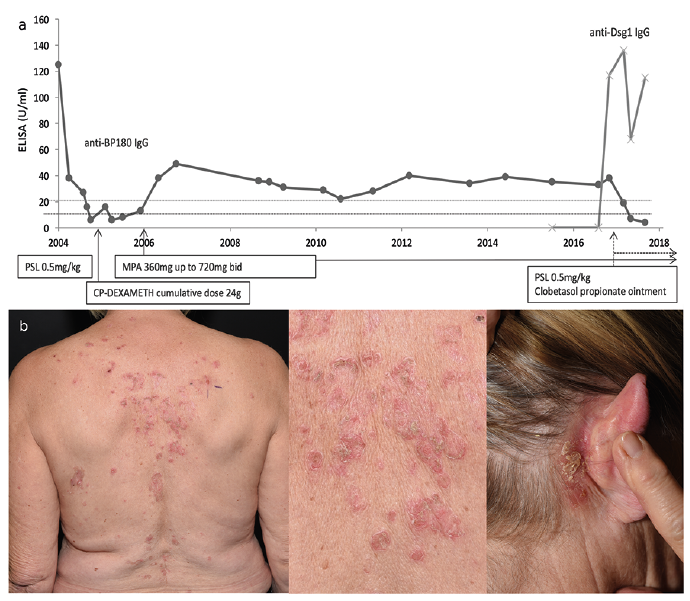
A 70-year-old woman had had severely itching erythematous plaques and newly occurring tense blisters for approximately 3 months before she consulted our department and was diagnosed with BP in 2004. The mucous membranes were uninvolved. Direct immunofluorescence (DIF) revealed linear IgG deposits at the dermo-epidermal junction zone (DEJZ), indirect immunofluorescence (IIF) on salt-split skin revealed IgG deposition at the blister roof. Cell surface antibodies were not detected in either direct or indirect immunofluorescence. The anti-BP180 NC16A enzyme-linked immunoassay (ELISA) (MBL, Nagoya, Japan) finally confirmed a diagnosis of BP. IgE levels were highly positive with 1,229 kU/l (normal range <100 kU/l). Her medical history was unremarkable, except for non-melanomic skin cancer. Prior to initiation of immunosuppressive therapy an underlying malignancy was excluded with various examinations, inclusive bone marrow puncture. Prednisolone was initiated at a dosage of 0.5 mg/kg body weight, resulting in clear improvement. However, when tapered to 10 mg per day, tense blisters developed again. In accordance with the dermatologist’s discretion, in 2004, treatment with cyclophosphamide-dexamethasone pulses and subsequent oral cyclophosphamide, with a cumulative dosage of 24 g was given for one year, without adequate clinical and serological control of disease and several recurrences. Only a treatment change to mycophenolic acid (MPA, dosage up to 720 mg bid) 1 year after BP diagnosis led to persistent clinical remission, while anti-BP180 ELISA remained positive during the following years (Fig. 1A).
In 2017, approximately 13 years after diagnosis of BP, the patient described new itching plaques on her trunk, rapidly followed by flaccid bullae, pustules and crusted erosions on her head, back and submammary folds (Fig. 1B). IgE levels were now within normal range. DIF revealed both linear IgG and C3c deposition along the DEJZ and epidermal cell surface IgG deposits, consistent with both a pemphigoid and pemphigus pattern. IgG1 and IgG4 deposits were found both along the DEJZ and the cell surface (Fig. S1a, b). In addition, a highly positive anti-desmoglein 1 (Dsg1) IgG ELISA and negative anti-desmoglein 3 (Dsg3) ELISA (MBL, Nagoya, Japan), as well as an anti-BP180 ELISA, confirmed the diagnosis of PF in coincidence with BP. Intense topical treatment and dosage escalation of MPA 360 mg tid lead to clinical remission within weeks. Some weeks later anti-BP180 ELISA was negative, while anti-Dsg1 ELISA decreased without serological remission (Fig. 1a).
Fig. 1. Longitudinal analysis of anti-BP180 IgG and anti-Dsg1 IgG enzyme-linked immunoassay (ELISA) titres with systemic treatment course and clinical picture of patient’s pemphigus foliaceus (PF). (a) The anti-BP180 ELISA titres stayed positive over the years, whereas they became negative after diagnosis of PF. The anti-Dsg1 ELISA was found positive approximately 13 years after diagnosis of bullous pemphigoid (BP). (Cut-off anti-BP180 ELISA <9 U/mL=negative, cut-off anti-Dsg1 ELISA <20 U/mL=negative). (b) Erythematous and crusted plaques and haemorrhagic erosions at the patient’s back and crusted erosions behind the right ear characteristic for PF (photographs taken 2017). PSL: prednisolone; CP-DEXAMETH: cyclophosphamide-dexamethasone; MPA: mycophenolic acid.
A few cases of transition from PF to BP (5–10) or the simultaneous existence of these conditions (11, 12) have been described previously. In most cases, shared histopa-thological and immunofluorescence findings were found. Clinical features indicated a diagnosis of either pemphigus or pemphigoid and, occasionally, false-positive reaction patterns were discussed (10). We report here a patient with clinical and serological transition of BP to PF after more than 13 years follow-up, which to our knowledge is the second case to be published (13). Our patient was in clinical remission from BP for several years, but showed persistent positivity for anti-BP180 IgG autoantibodies, possibly caused by the presence of long-lived memory B cells, i.e. long-lived plasma cells, a phenomenon which has been proven to be important for life-long immunity against infectious diseases (14). For a short time, the patient developed dual features of BP and PF in DIF, IIF and ELISA diagnostics, whereas at this point the clinical phenotype resembled PF. Dose escalation of MPA was accompanied by serological transition, and anti-BP180 ELISA became negative within weeks. Retrospective evaluation of patient’s blood samples prior to clinical onset of PF disclosed negative anti-Dsg1 IgG as well as anti-BP230 IgG autoantibodies. Evaluation of IgG subclasses showed deposition of both IgG1 and IgG4, with IgG1 more pronounced along the DEJZ, whereas IgG4 was predominantly found at cell surface and increased over time (Fig. S1b).
The development of independent and non-cross-reactive autoantibodies is thought to be caused by diversification and modification of an auto-aggressive T-cell response to secondary epitopes in the course of persisting inflammatory cascades and tissue damage induced by the dominant self-epitope (2–4, 15). In our case, detection of BP antibodies was evident over a period of 13 years, which is in line with the above-mentioned concept of ongoing serological active disease. Sárdy et al. hypothesized that insufficient immunosuppressive treatment with ongoing serological activity might facilitate ES, and leads to a more complex clinical course (16). Further studies are needed to investigate whether B-cell depletion therapies (e.g. using rituximab) in patients with extensive involvement in autoimmune blistering disorders, in particular patients with long-lasting autoantibody positivity could prevent ES and a prolonged clinical course.
The authors thank the patient. We also thank Annegret Bedorf for excellent technical assistance. DK’s work has been supported by EB research Partnership and the Mathilde-Wagner-Habilitationspreis from the University of Freiburg.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize