


1Faculty of Health Sciences, University of Southern Denmark, 2Department of Dermatology and Allergy Centre, Odense University Hospital, and 3Department of Pathology, Odense University Hospital, Odense, Denmark
Hereditary leiomyomatosis and renal cell cancer is a genodermatosis with an autosomal dominant inheritance pattern. It is a tumour predisposition syndrome characterized by cutaneous and uterine leiomyomas, and increased susceptibility to develop renal cell carcinoma. There are 200–300 families with hereditary leiomyomatosis and renal cell carcinoma reported worldwide, but the syndrome is believed to be underdiagnosed. Cutaneous leiomyomas are small smooth muscle tumours that tend to grow over time. Larger lesions, in particular, can cause pain or itching. Uterine leiomyomas have a high penetrance in women with hereditary leiomyomatosis and renal cell cancer. They frequently cause symptoms, and surgical intervention is often necessary. Hereditary leiomyomatosis and renal cell cancer-associated renal cell carcinomas have a high potential to metastasize. Patients are diag-nosed by genetic testing if a pathogenic mutation is demonstrated in the gene encoding fumarate hydratase. Immunohistochemistry may be a useful diagnostic approach in patients without a detectable pathogenic mutation. Diagnosed patients should be monitored for renal tumours in a lifelong surveillance programme.
Key words: hereditary leiomyomatosis; cutaneous leiomyomas; uterine leiomyomas; renal cell carcinoma; cancer surveillance.
Accepted Oct 28, 2019; E-published Oct 30, 2019
Acta Derm Venereol 2020; 100: XX–XX.
Corr: Anders Würgler Hansen, Medical Student, Faculty of Health Sciences, University of Southern Denmark, Kloevervaenget 24A 2, DK-5000 Odence C, Denmark. E-mail: anders.wurgler.hansen@rsyd.dk
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is an inherited tumour predisposition syndrome. Around 75% of individuals with HLRCC develop cutaneous leiomyomas, which are skin tumours that can cause pain and itching. Most women with HLRCC develop uterine leiomyomas that often cause gynecological symptoms and typically require surgery. Around 20% develop renal cell carcinoma. HLRCC-associated renal cell carcinomas are aggressive tumours with a high potential to metastasize. Individuals with features suggestive of HLRCC and at-risk family members should undergo genetic testing. Individuals harbouring a pathogenic mutation should be offered lifelong surveillance so renal neoplasms are detected and treated in time.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a rare genodermatosis with an autosomal dominant inheritance pattern, caused by inherited or de novo mutations in the gene encoding fumarate hydratase (FH) (1). HLRCC is a tumour predisposition syndrome characterized by the propensity to develop single or multiple cutaneous leiomyomas (CLM), uterine leiomyomas (ULM), and renal cell carcinomas (RCC). Blum & Jean (2) linked leiomyomas of the skin with uterine leiomyomas for the first time in 1954. In the literature, the syndrome is referred to as Reed’s syndrome, multiple cutaneous and uterine leiomyomatosis (MCUL), and leiomyomatosis cutis et uteri, amongst other names (3). The eponym Reed’s syndrome dates back to 1973, when Reed and colleagues noticed an accumulation of the condition in 2 families with an autosomal dominant inheritance pattern (4). The syndrome was subsequently renamed HLRCC in 2001, when the condition was linked with an increased susceptibility to certain subtypes of RCC (5).
The exact prevalence of HLRCC is unknown; however, 200–300 families have been reported in the literature (1, 3). Mostly Western-European, Finnish and North-American families have been reported (6–8). Gardie et al. reported 44 families with confirmed HLRCC in France (9). Toro et al. reported 35 HLRCC families in North America (7), where Wei et al. (10) later reported an additional 21 families. Lehtonen et al. (6) reported 7 Finnish families with HLRCC. Reports of Colombian, Japanese, Indian, Chinese and African-American HLRCC families suggest that the syndrome occurs worldwide (10–14). HLRCC is believed to be an underdiagnosed syndrome, and the actual number of HLRCC families is likely to be higher (8).
The genetic predisposition to CLM, ULM and RCC is caused by mutations in the FH-gene on chromosome 1q (5). HLRCC is an autosomal dominant disease with sporadic cases reported (8, 15).
FH acts as a tumour suppressor gene in accordance with Knudson’s 2-hit hypothesis (5, 16). Tumour tissue displays biallellic FH inactivation where an acquired somatic FH mutation can be demonstrated in addition to the inherited germline mutation (17, 18). An excess of 200 unique potential pathogenic FH variants have been reported (19). The gene product is the enzyme fumarate hydratase or fumarase, which catalyses the conversion of fumarate to malate in the tricarboxylic acid cycle (TCA) (20). An inactivating mutation, most commonly a missense mutation of the wild type allele, leads to decay of the mRNA or truncation of the gene product, resulting in an either markedly reduced or absent enzyme activity (7, 10, 21–23). Fumarate accumulates in cells in the absence of FH (24). The mechanism leading to tumourigenesis is poorly understood, although hypoxia inducible factors (HIF) in the hypoxia path-way are hypothesized to play a role. Fumarate inhibits HIF prolyl hydroxylase (HPH), an enzyme that catalyses the degradation of HIF, leading to elevated intracellular levels of HIF (25). HIF-1α, HIF-2α and HIF-1β promotes the transcription of target genes involved in angiogenesis, cell proliferation and cell survival (26). Direct sequencing reveals a pathogenetic FH germline mutation in 71–100% of families with clinically suggestive HLRCC (3, 8, 9, 18). In cases without detectable pathogenic FH mutations, a reduced or absent FH activity may be detected in tumour tissue by a biochemical assay (22, 27, 28). Whole-gene deletions, exon deletions and large rearrangements have rarely been demonstrated in HLRCC individuals without detectable FH mutations (3, 9, 20, 21, 29).
HLRCC displays a broad clinical spectrum, as some cases with FH mutations are asymptomatic while others have severe cutaneous, uterine or renal manifestations (9, 21). The penetrance of CLMs has been reported to be higher in men with FH mutations compared with women, as a study reported that CLMs were present in all men by age 35 years, while being present in only 55% of women (6). Predicting the phenotype based on a specific inherited gene variant is not possible (1, 9, 23). Due to recruitment of individuals based on positive findings in studies and publications of HLRCC, the prevalence of clinical manifestations might be overestimated (7, 8, 10).
Cutaneous leiomyomas
CLMs are the most frequent manifestation of disease leading to a diagnosis (8). They are benign, firm, smooth, skin-coloured to erythematous or hyperpigmented papulonodules (3, 8, 30) (Figs 1 and 2). CLMs range in size between 2 and 20 mm in diameter (3, 8). Histologically, CLMs are subdivided into piloleiomyomas, genital leiomyomas and angioleiomyomas. Piloleiomyomas, being the most frequent in relation to HLRCC, emerge from the arrector pili muscles of hair follicles (Fig. 3) (3). Angio-leiomyomas are derived from the vascular smooth muscle tissue in the tunica media of blood vessels (31). Genital leiomyomas originate from the dartoic smooth muscle tissue of the vulva, scrotum and nipple-areola complex (31, 32). While angioleiomyomas and genital leiomyomas are both rare, the former is often painful and the latter painless (3, 31). Piloleiomyomas can be multiple or solitary (3, 8). They are usually located on extensor surfaces of the trunk, upper/lower extremities, shoulders and, less frequently, on the face and neck (3, 8, 30). Multiple lesions can be described as clustered or scattered, while segmental, zosteriform, linear, and disseminated distributional patterns have also been reported (3, 8, 30). Tumors are usually bilateral, but can be unilateral or symmetrical (3, 8). CLMs appear in 68–84% of HLRCC patients, and up to 100% of HLRCC families (9, 10, 15, 21). In 9–15% of individuals they are the only clinical manifestation of the condition (7, 8, 15, 23). They initially appear in the 2nd to 4th decade, with a mean age at onset of 24–30 years, although they have been reported in a patient as young as 9 years of age (7, 8, 23). Approximately 75–90% of HLRCC individuals experience variable itching or pain from their skin lesions, which may be provoked by direct contact, emotions, or fluctuations in temperature or pressure (3, 7, 8, 30). The number of CLMs varies widely among affected individuals, ranging from a single lesion to more than 100 (7, 8). Lesions generally increase in number and size over time, and especially larger lesions tend to be symptomatic (15, 21). Individuals with CLMs have reported a moderate to severe negative impact on their quality of life caused by their skin lesions (8). The potential of cutaneous and uterine leiomyomas to transform into leiomyosarcomas is not well established. Only 6 cases of cutaneous leiomyosarcomas have been reported in FH mutation carriers (7, 10, 23, 33, 34), 2 of which presented with rapid growth of an existing CLM (23). Similarly, uterine leiomyosarcomas have rarely been linked to HLRCC (4–6, 35). Leiomyosarcomas are difficult to distinguish clinically from CLMs, but may be larger, ulcerated, irregularly shaped and increasingly painful (3). Histopathological examination of tumour tissue is required for a firm diagnosis (36). These tumours rarely metastasize (3). Although speculative, looser diagnostic criteria of leiomyosarcomas in the past may have led to uterine and cutaneous leiomyomas being mistaken for leiomyosarcomas (23).
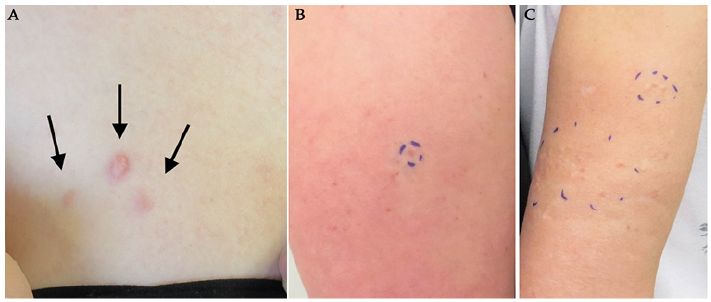
Fig. 1. Presentations of cutaneous leiomyomas. (A) Small cluster of reddish cutaneous leiomyomas (CLM) with 1 prominent lesion and 2 surrounding smaller lesions located on the upper trunk of a 35-year-old woman with genetically verified hereditary leiomyomatosis and renal cell cancer (HLRCC). (B) A single CLM on the upper arm of a woman with HLRCC. (C) Clusters of CLMs in the woman’s mother.
Fig. 2. Surgical excision of cutaneous leiomyomas on the chest of a 20-year-old patient under current investigation for hereditary leiomyomatosis and renal cell cancer. (A) Cutaneous leiomyomas prior to surgical excision. (B) Keloid formation following surgical excision.
Fig. 3. Immunohistochemical staining of a cutaneous leiomyoma in a hereditary leiomyomatosis and renal cell cancer patient. Tumour cells are fusiform with small clear uniform nuclei. The tumour tissue predominantly consists of smooth muscle cells with positive staining for smooth muscle markers alpha-smooth muscle actin (ASMA), desmin and vimentin. (A) Haematoxylin and eosin staining of cutaneous leiomyoma. (B) ASMA staining of cutaneous leiomyoma. Original magnification x40.
Uterine manifestations
Uterine fibroids range in number between 1 and 20, and in diameter between 1 and 10 cm (7, 10). HLRCC-associated ULMs are diagnosed in the 2nd to 5th decade, with a mean age of 30 years (6–8, 10). ULMs occur in 73–100% of HLRCC families and in 76–100% of women with HLRCC (7–10, 15, 21, 23). In 7–14% of cases, ULMs are the only disease manifestation (8, 10). The mean age at diagnosis of ULMs is 28–30 years, ranging from 18 to 53 years (7, 8, 10, 37). In the general public, the prevalence of symptomatic uterine fibroids is 9% (38), and they are diagnosed approximately 10 years later than in women with HLRCC (37). The majority of women with HLRCC-associated ULMs experience gynaecological symptoms, most commonly dysmenorrhea, followed by menorrhagia and irregular menses (7, 8, 21, 37). Women with HLRCC often experience gradual worsening of these symptoms prior to diagnosis (8). HLRCC-associated ULMs are associated with female infertility characterized by difficulty achieving conception and occurrence of miscarriages (8). Up to 91% of women with a pathogenic FH mutation undergo surgical intervention, such as hysterectomy or myomectomy, at a mean age of 35–36 years (7, 10, 23), and many undergo surgery before the age of 30 years (7, 10). HLRCC-associated ULMs are associated with various degrees of negative impact on quality of life (8).
Renal manifestations
HLRCC-associated RCC display a broad spectrum of architectural growth patterns, including papillary, tubulo-papillary, tubular, solid and cystic elements (10, 23, 39–41). The syndrome mainly predisposes to RCCs of a type 2 papillary morphology (PRCCII), but tumours of collecting duct, clear cell, sarcomatoid and oncocytic origin have also been reported (7, 9, 22, 23, 40). RCCs are predominantly solid and unilateral, but can also be multifocal and bilateral (10, 14, 40, 42). Tumour cells have large nuclei, with prominent inclusion-like eosinophilic nucleoli, surrounded by a perinuclear clear halo of cyto-plasm (40, 43). Renal tumours vary in size between 2 and 22 cm in diameter (5, 7, 8). RCCs usually display an aggressive growth pattern, with invasion of nearby tissue, and a high potential to metastasize (14, 23, 44). Symptoms include haematuria, flank pain, lower back pain, palpable mass and symptoms from metastases (3). Patients can debut with RCC as the only clinical manifestations of the syndrome (10, 45). HLRCC-associated RCCs have a poor prognosis, with a 5-year survival of 31%, as reported by Toro et al. (7). Furthermore, Muller et al. reported a median survival of 18 months for metastatic disease (23). RCC is diagnosed in approximately 14–62% of HLRCC families (7, 9, 10), and in 16–24% of individuals with HLRCC (7, 9, 10, 23). However, a Dutch study found that only 6% of patients with HLRCC were diagnosed with RCC (21). The mean age at diagnosis of HLRCC is 39–46 years (range 10–90 years) (6–9, 14, 41, 46, 47), approximately 20–25 years earlier than the diagnosis of sporadic RCC in the general European and American population (48, 49).
The need for treatment of CLMs is individual and depends mainly on pain and cosmetic appearance of the lesions. Conservative approaches involve avoiding symptomatic triggers, such as direct contact and changes in temperature (3). Issues of cosmetic appearance might be solved by choice of clothing and use of make-up (3). CLMs can readily be removed by standard excision, electrodessication, CO2 laser ablation or cryotherapy (3, 21). The choice of surgical intervention depends largely on the preferences of the clinician. However, the indication for cosmetic removal of CLMs should be weighed against the risk of scarring and keloids (Fig. 2).
Topical treatment by lidocaine or botulinum toxin may offer pain relief in symptomatic lesions (3). Systemic pharmacological treatment with nitroglycerine, nifedipine, gabapentin, phenoxybenzamine, and doxazocin is sometimes attempted to relieve pain (3, 21, 50).
Gonadotropin-releasing hormone agonists and progesterone-releasing intrauterine devices can be used as medical treatment for ULMs (3). Surgical interventions are most commonly myomectomy and hysterectomy. Myomectomy is a uterus-preserving surgical intervention, with removal of the fibroids from the uterus (50). Other surgical interventions include uterine artery embolism and electrosurgery (3, 8, 10). Patients are counselled on family planning prior to choice of treatment (21).
Due to the often aggressive growth pattern of HLRCC-associated RCCs, surgical intervention is recommended even in small tumours, with extirpation of all neoplastic tissue and possibly retroperitoneal lymph node dissection (47). If possible, nephron-sparing surgery with partial nephrectomy is preferred in order to preserve renal function (47). There is no standard treatment option for metastatic disease, and very limited treatment data avail-able (51). Medical treatment options usually interact with components of the mammalian target of rapamycin (mTOR) pathway and the HIF pathway, including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) (47, 51). The most commonly prescribed drugs targeting these pathways are bevacizumab, temsirolimus, everolimus, pazopanib, axitinib, sunitinib, sorafenib and erlotinib (47, 51). These agents may be used in combination, in order to achieve the best response (47, 51, 52). Choice of treatment modality depends largely on patient preferences and comorbidities (3). Bevacizumab in combination with erlotinib showed promising results in the treatment of metastasized HLRCC-associated RCC (51, 52). There is currently ongoing research of the use of inhibitors of glycolysis in the treatment of HLRCC-associated RCC, as the tumour cells are dependent on glycolysis for ATP production due a TCA cycle defect (47, 53). However, such therapies have not yet proven effective (54).
A diagnosis of HLRCC is confirmed through genetic testing (50). Routine molecular screening of ULM tissue has been suggested (55, 56). Random inactivation of FH is a commonly seen phenomenon in sporadic ULMs (57, 58). This is due to somatic biallelic inactivation rather than germline mutations (57, 58). Therefore, screening of ULM tissue for FH-deficiency was deemed impractical in individuals without features suggestive of HLRCC (57, 59). Pathological screening of ULM morphology was recently proposed to trigger genetic testing, in cases suggestive of FH deficiency (60). This strategy might help to detect women eligible for genetic counselling (60).
Immunohistochemical (IHC) examination of neoplasms associated with HLRCC can support the diagnosis in cases where a pathogenic germline mutation is not known (47, 59, 61). In addition, IHC examination plays an important role in differentiating HLRCC-associated RCC from other high-grade RCCs (39, 41, 59, 61). Alterations of succinate dehydrogenase (SDH), an acid cycle enzyme linked to renal tumors, cannot be demonstrated by IHC staining of FH-deficient tumor tissue (62). However, loss of FH expression in tumour tissue can be identified by absent FH staining (59). S-(2-succinyl) cysteine (2SC) is another highly specific IHC biomarker in detecting HLRCC-associated RCC (39, 41, 59). 2SC is produced in a process termed protein succination, wherein high levels of fumarate interact with cysteine sulphydryl groups of cellular proteins to form a stable chemical modification (61). Absence of FH staining and/or presence of 2SC staining in tumour tissue is strongly correlated with HLRCC (39, 41, 59, 61, 63). While IHC examination alone cannot replace genetic testing, it may contribute to weakening or strengthening the suspicion of HLRCC (62, 64). Immunostaining of CLMs has been suggested as a routine screening tool for HLRCC (62, 65, 66). However, Harvey & Wood argue that, compared with immunostaining techniques, the very presence of multiple CLMs is more cost-effective and has a higher sensitivity for an underlying germline mutation while the specificity is unestablished (67). A revised proposal for diagnostic criteria of HLRCC is shown in Table I (1, 3, 9, 21, 50).
Table I. Proposed diagnostic criteria for hereditary leiomyomatosis and renal cell cancer (HLRCC)
The differential diagnostic considerations depend on the clinical findings. CLMs are rare and their presence should always lead to a suspicion of HLRCC (1, 43, 68). A large number of skin tumours may cause pain (69). The most relevant differential diagnostic considerations to painful CLMs are shown in Table II (3, 30, 69–71). Although the clinical spectrum of CLMs is broad, many of these tumours can readily be eliminated as likely differential diagnoses based on the clinical findings. A biopsy is required for a firm diagnosis (30).
ULMs are frequent in the general population and constitute the most common pelvic tumour in women (72).
The main differential diagnoses in relation to renal tumours are the autosomal dominant renal cancer syndromes: hereditary papillary renal cancer (HPRC), Von Hippel-Lindau syndrome (VHL) and Birt-Hogg-Dube syndrome (BHDS) (73). Predisposition to papillary type 1 RCC is the only manifestation of HPRC (1). VHL is characterized by clear cell RCC and renal cysts (74). Other common findings include central nervous system haemangioblastoma, pancreatic tumours and cysts, and pheochromocytoma among others (74). VHL can be differentiated from HLRCC by the lack of cutaneous and uterine lesions (1). BHDS is associated with lung cysts, that may erupt and cause pneumothorax, and a triage of skin lesions, including fibrofolliculomas, trichodiscomas and acrochordons (skin tags) (75). Distinctive from CLMs, fibrofolliculomas and trichodiscomas are painless, pale, millimetre small lesions, and are mainly located on the face, neck and upper torso (75). The renal tumours seen in BHDS constitute in a wide histological spectrum. Patients most often present with hybrid tumours, a combination of chromophobe RCC and renal oncytoma, but also chromo-phobe RCCs, clear cell RCCs or renal oncocytomas are frequently associated with BHDS (75). BHDS is not associated with an increased frequency of ULMs (1).
Table II. Clinical differential diagnoses of painful cutaneous leiomyomas
There is a lack of consensus regarding a surveillance programme of pathogenic FH-mutation carriers (30). Al-though suspected, a firm association of cutaneous leiomyosarcomas with HLRCC has not been established (23). We recommend a thorough dermatological examination every second year, starting from the onset of CLMs (Table III). Patients should be instructed to seek medical attention in case of rapid growth in skin lesions. Annual ultrasonic gynaecological examinations starting at the age of 20 years, in FH mutation carriers, is recommended in order to diagnose and monitor asymptomatic ULMs (Table III) (3, 21, 50). Prevention of RCCs is a major focus of the surveillance programme (21). Renal surveillance by annual contrast-enhanced magnetic resonance imaging (MRI) is proposed to start at the age of 10 years by Patel et al. (3), and at the age of 8 years by Schmidt & Linehan (50). Due to numerous reports of HLRCC-associated RCC in individuals younger than 20 years of age (8, 40, 46, 76, 77), the youngest reported at age 10 (30), we propose annual MRI scans starting at age 10 years (Table III). While renal ultrasound has been suggested to play a role in assisting MRI in the surveillance programme (21), Schmidt & Linehan (78) does not recommend this modality for routine screening of HLRCC-associated renal tumours. Furthermore, family members of patients with proven HLRCC should undergo early genetic testing in order to be included in the surveillance programme at an early age (1, 78).
Table III. Proposed surveillance programme of pathogenic fumarate hydratase mutation carriers
The prognosis of HLRCC depends first and foremost on the detection of HLRCC-associated renal cancers. It is imperative to diagnose the syndrome early, and include patients in an aggressive surveillance programme, so that renal tumours are diagnosed and treated in time.
At-risk family members and offspring of confirmed FH-mutation carriers should undergo genetic testing in order to identify the necessity for inclusion in the surveillance programme. HLRCC patients and at-risk family members should be offered genetic counselling and be provided with information regarding the syndrome (1). FH mutation carriers should ideally receive genetic counselling before starting a family, in order to discuss the following prior to pregnancy; fertility issues, prenatal testing options, and risk-assessments with regards to passing on the pathogenic gene variant to offspring (1).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize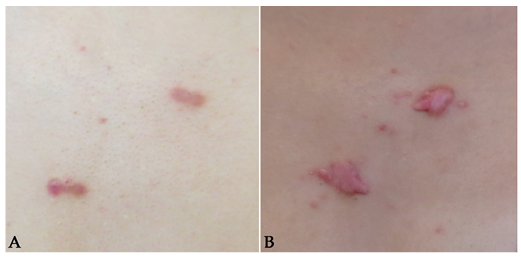 Click to show fullsize
Click to show fullsize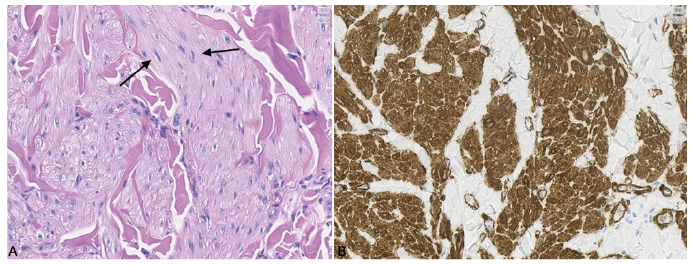 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize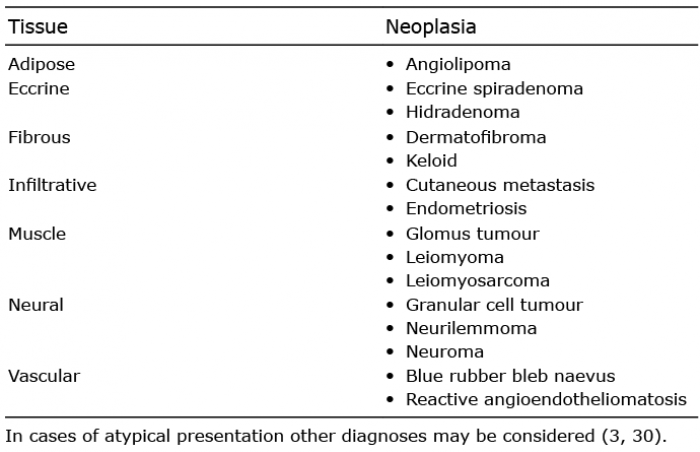 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize