


Dokumentationszentrum schwerer Hautreaktionen (dZh), Department of Dermatology, Medical Center and Medical Faculty, University of Freiburg, Freiburg, Germany
Bullous drug eruptions are infrequent, but because they pose a challenge both to affected patients and to treating physicians they are considered to be the most severe cutaneous adverse reactions (SCAR). It is important to recognize these conditions and to differentiate them from other clinical entities involving blister formation. There may be early signs and symptoms that indicate a severe bullous drug eruption even before blisters and erosions of the skin and mucous membranes become obvious. Once the diagnosis is suspected, appropriate diagnostic procedures and adequate management must be initiated. The latter includes identification of the potentially inducing drug, although it should be taken into account that not all cases of bullous eruptions are drug-induced. In cases with drug causality the potentially culprit agent must be withdrawn, while in cases with other aetiology the underlying condition, e.g. an infection, must be treated appropriately. In addition to best supportive care, immunomodulating therapy may be considered.
Key words: severe cutaneous adverse reaction; Stevens-Johnson syndrome; toxic epidermal necrolysis; generalized bullous fixed drug eruption.
Accepted Jan 24, 2020; Epub ahead of print Feb 6, 2020
Acta Derm Venereol 2020; 100: adv00057.
Corr: Maja Mockenhaupt, Dokumentationszentrum schwerer Hautreaktionen (dZh), Department of Dermatology, Medical Center and Medical Faculty, University of Freiburg, Hauptstr. 7, DE-79104 Freiburg, Germany. E-mail: dzh@uniklinik-freiburg.de
Drug reactions with blisters (known as bullous drug reactions) are challenging for patients and physicians. Often there are early signs and symptoms that may lead to the suspicion of a bullous drug reaction before blisters and erosions of the skin and mucosa appear. Once the diagnosis is suspected, appropriate diagnostic and therapeutic procedures must be initiated. A detailed history, including clinical symptoms, drug use and infections, is crucial. In cases with drug causality, the potentially culprit agent must be withdrawn, while in cases with other aetiology, the underlying condition, e.g. infection, must be treated. In addition to best supportive care, immunomodulating therapy may be considered.
Bullous drug reactions generally occur as a result of medication use, but there are also other possible causes. One of the major challenges is to identify at a very early stage whether the reaction will be severe and life-threatening. Once blisters are present, differentiation between types of reaction, such as Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), generalized bullous fixed drug eruption (GBFDE) and, sometimes, bullous autoimmune disease, is also challenging, since, for example, conditions such as GBFDE or IgA-linear dermatosis can mimic SJS/TEN. Differentiation is important as prognosis and treatment modalities differ substantially.
In 1922, the American paediatricians Stevens & Johnson (1) reported 2 cases of a disseminated cutaneous eruption associated with erosive stomatitis and severe ocular involvement. In 1956, the Scottish dermatologist Lyell (2) described patients with epidermal loss secondary to necrosis, and introduced the term “toxic epidermal necrolysis”. However, Lyell did not refer to the findings of Stevens & Johnson at that time, but in a later reappraisal evaluated the original 4 cases in his publication as SJS/TEN, staphylococcal scalded skin syndrome (SSSS) and generalized bullous fixed drug eruption (GBFDE) (3). The histopathological difference between an intraepidermal subcorneal separation in one case had already been described in his first publication, but it took until 1971 to identify a staphylococcal exotoxin as the cause of this reaction and to name it accordingly (4). Around the same time Kauppinen (5) from Finland separated a multilocular or GBFDE from SJS and TEN through clinical features and behaviour in allergological testing.
Over the years, due to similarities in clinical and histopathological features, SJS and TEN have been included in the spectrum of erythema multiforme (EM), which was first described by von Hebra in 1860 (6). However, several attempts have been made to disentangle, regroup and rename the reactions. Ruiz-Maldonado (7), for example, proposed the term “acute disseminated epidermal necrosis” for SJS, TEN and “transmission of forms”, but did not separate EM, whereas Lyell (8) suggested the name “exanthematic necrolysis” for SJS/TEN. Based on the original descriptions and the observation that SJS may progress into TEN, an international group of dermatologists developed a consensus definition that separates these conditions from EM. Because SJS and TEN share a clinical pattern, histopathological findings, aetiology, risk factors, and mechanisms, they are considered as severity variants of a single disease entity that differs only in the extent of skin detachment related to the body surface area (BSA) (9). Therefore, it seems more appropriate to use the term “epidermal necrolysis” or “epithelial necrolysis” (referring to skin and mucosa) for both (10).
Epidermal necrolysis (EN) is a rare condition with an overall incidence of 1–2 cases per million persons, estimated using strictly validated cases of a prospective population-based registry (11, 12). However, incidences as high as 5–6 cases per million per year derive from medical databases not primarily designed for epidemiological analysis of rare diseases (13). EN can occur at any age, but the risk increases with age and the highest incidence is seen in elderly persons over 65 years of age (14). The mean age of patients was 53.4 years (range 1–94 years) in a cohort of more than 2,200 patients (15). Women are more frequently affected, with a sex ratio of 0.6. Patients infected with human immunodeficiency virus (HIV) and, to a lesser degree, patients with collagen vascular disease (also called connective tissue disease, including rheumatoid arthritis, systemic lupus erythematosus, Sjögren syndrome, dermatomyositis, polymyositis, scleroderma, mixed connective tissue disease and some types of vasculitis) and cancer are at increased risk (11, 16). The overall mortality associated with EN is 22–25%, varying from approximately 10% for SJS to almost 50% for TEN (17–19). Several factors contribute to poor prognosis, such as larger extent of skin detachment, older age, and underlying comorbidity.
In contrast, the mortality for erythema (exsudativum) multiforme majus (E(E)MM; i.e. EM with mucosal involvement) is very low, affecting few individuals of older age and underlying conditions. The majority of patients are young (80% are younger than 40 years, 45% are under 18 years) and male (approximately 75%) (9, 20). The incidence of cases of severe EMM leading to hospitalization is of approximately the same order of magnitude as that of EN (SJS-TEN), with milder cases (EM minus with only skin involvement or cases with only mucosal involvement) occurring more frequently (15, 20).
To date, estimates of the incidence of GBFDE are lacking, since there are currently no population-based data. As with most types of cutaneous adverse reactions, GBFDE more frequently affects women. Of the affected patients 70% are older than 70 years and approximately 22% of patients die due to advanced age and disease severity (21).
EN is characterized by erythematous skin, epidermal detachment and erosions of mucous membranes. The erythematous exanthema consists of atypical flat target lesions (these lack the typical 3-zone, target-like constellation of so-called typical target lesions seen in EM) and/or macules that frequently tend to become confluent and spread from cranial to caudal. Blisters develop on the erythema and coalesce. Usually, at least one mucous membrane is affected by erosion in addition to the skin. Fever and malaise are very common (10). The condition is classified according to the consensus definition: skin detachment of less than 10% of the BSA refers to SJS, and more than 30% of the BSA to TEN. Skin detachment between these values is defined as SJS/TEN-overlap (Table I, Fig. 1) (22). In approximately 95% of cases, haemorrhagic erosions of mucous membranes, including eyes, lips, mouth, vulva, glans penis, and sometimes also trachea, bronchi, urethra and anus, are present (Fig. 2). Due to the fact that the skin detachment progresses, turning a case initially thought of as SJS into TEN, and due to the fact that SJS and TEN share the same aetiology and pathogenesis, they are considered as a single disease entity of different severity (9).
Table I. Consensus definition of epidermal necrolysis (EN) (22)
Fig. 1. Confluent macules with confluent blisters, leading to large areas of skin detachment in epidermal necrolysis (patient’s back).
Fig. 2. Haemorrhagic erosions of mucous membranes in epidermal necrolysis or erythema multiforme majus: (a) blepharitis, (b) erosions of lips and oral mucosa, genital erosions in (c) a male and (d) a female patient.
Due to the same type of mucosal involvement, EM with mucosal involvement (erythema multiforme majus; EMM) was assumed to be a less severe form of SJS. However, this incorrect classification may lead to false assessment of causal factors, which in SJS/TEN are predominantly medications and in EMM almost exclusively infections (9, 10, 20). Furthermore, younger patients with EMM may be severely ill with high fever and overall poor general state of health (20).
EMM and SJS can generally be well differentiated based on the consensus definition (in more than 90% of cases), especially when typical targets on the limbs are present (Fig. 3). However, differentiation may be challenging in the case of atypical EMM involving atypical “giant targets”. This also accounts for the mainly truncal and generalized distribution of typical target lesions, especially in children and adolescents, since these lesions sometimes coalesce. The description of a typical and atypical type of EMM helps to better classify the various patterns of EM and their distinction from SJS (23). Moreover, due to their demarcation towards intact skin, older “giant targets” may resemble resolving patches in GBFDE (18).
Besides EMM, GBFDE is an important differential diagnosis of EN. This reaction is typically characterized by well-defined round or oval, egg-sized patches of dusky violaceous or brownish colour. Blisters may develop on these patches, but the skin remains intact between the areas of blistering and, in most cases, skin detachment does not affect more than 10% of the BSA (Fig. 4). However, the reaction may also present with diffuse erythema and blisters, which will show demarcation during the course. There is a debate among experts as to whether the rare cases of TEN on large erythema are potentially severe forms of GBFDE (10, 24).
Patients with GBFDE usually do not develop fever and malaise, but there may be mild mucous membrane involvement, with the genital and/or oral mucosa affected, but not the ocular surface. Milder eruptions are frequent in the patient’s medical history (18, 21, 24).
To supplement the consensus definition for EN described above (22), the RegiSCAR-group developed a score for the diagnostic differentiation of GBFDE, which is currently in the validation phase and has not yet been published. There are no specific laboratory parameters to differentiate between the various types of blistering reactions.
Fig. 3. Typical target lesions with central blisters in erythema multiforme majus (on the leg).
Fig. 4. Well-demarcated erythematous patches with blisters in generalized bullous fixed drug eruption (on the back).
The histology of EN reveals necrotic (dyskeratotic or apoptotic) keratinocytes, either in a disseminated distribution or as complete epidermal necrosis with subepidermal blister formation. Localization and timing of sample collection are important: if the biopsy is taken from the central blister of an EMM target, complete epidermal necrosis may also be visible, as well as a sparse superficial lymphocytic infiltrate in the dermis, often in a perivascular location (25, 26). Therefore, histopathology can only confirm the clinical condition within the spectrum of disease but is unable to proof the specific clinical form. The same accounts for the histology of GBFDE, in which a distinction is sometimes possible in the course of the disease. If a biopsy is taken at a later stage, a deep perivascular infiltrate containing neutrophils and eosinophils may be seen, and potentially also pigment deposits (26, 27).
EN and GBFDE must be differentiated from staphylococcal scalded skin syndrome (SSSS), which histologically shows intraepidermal, subcorneal separation (4). Bullous autoimmune dermatoses, such as bullous pemphigoid, linear IgA dermatosis, pemphigus vulgaris, and paraneoplastic pemphigus, should be included in the differential diagnosis (10). Therefore, if one of these diseases is suspected, a direct immunofluorescence test as well as serological autoimmune parameters (e.g. anti-BP 180-, 230-, desmoglein antibodies) should be performed (10). Linear IgA bullous dermatosis (LABD) can imitate SJS/TEN, as has been described in several case reports and case series (28, 29). Some authors reported a more severe pattern with larger areas of skin detachment in cases that were drug-induced, with vancomycin being a frequent cause (29). Other disorders that should be considered in the differential diagnosis include widespread drug eruptions, erythroderma, exfoliative dermatitis, and subacute cutaneous lupus erythematosus (10, 18). Acute generalized exanthematous pustulosis (AGEP) may mimic EN, when confluence of pustules appears to reveal a positive Nikolsky’s sign. However, AGEP does not turn into EN, since there is no primary epidermal necrosis, but there is rapid healing of the subcorneal lesions. Bullae may occur in body areas with oedema, leading to widespread intraepidermal blister formation and secondary necrosis of the blister roof (30). If tension blisters appear due to oedema in drug reaction with eosinophilia and systemic symptoms (DRESS), EN might be suspected, but early histopathology will show that there is no full-thickness necrosis leading to epidermal detachment and that the subepidermal separation occurs first followed by secondary necrosis of epidermal cells (18). In addition, atypical target lesions on the limbs and erosions of the lips may raise the suspicion of EN, although features such as facial oedema and erythema with inflammatory infiltration of the skin point to DRESS. Therefore, it is important to monitor specific laboratory values relevant for a diagnosis of DRESS, e.g. eosinophilia, liver enzymes, kidney parameters, etc. Liver involvement, indicated by at least a 2-fold increase in transaminases, on 2 different days may occur when eosinophilia has already turned to normal values. When the skin eruption heals, widespread post-inflammatory desquamation is frequently observed and sometimes mistaken for skin detachment in EN (18, 31).
Other differential diagnoses vary with the clinical pattern and during the course of the reaction. In the early stage of the disease, maculo-papular, multiforme- or target-like drug eruptions, which can also present with oral lesions and conjunctivitis, must be considered, especially in elderly patients (Table II) (32). Varicella and other viral exanthems are important differential diagnoses when the first signs and symptoms occur in children (10, 18, 33).
Table II. Differential diagnoses of epidermal necrolysis (EN) (10)
EN typically begins with unspecific prodromal symptoms, such as sore throat, runny nose, cough, headache, fever, and malaise, preceding mucocutaneous lesions by 1–3 days. These symptoms are followed by the appearance of erythematous macules and atypical targets of the skin that may be confluent and on which blisters occur. Burning or stinging of the eyes, and pain when swallowing or urinating, develop progressively, heralding mucous membrane involvement. Most reactions start with non-specific symptoms, followed either by cutaneous or mucosal involvement, but some may begin directly with specific lesions of the skin and mucous membranes. The rapid progression of such symptoms, the addition of new signs, severe pain, and rapid decline in the patient’s general state of health should prompt the suspicion of a severe disease (10, 33).
In the majority of EN-cases the eruption initially shows a symmetrical distribution on the face, the upper trunk, and the proximal parts of the limbs. The distal parts of the arms and legs are often spared, but the eruption may extend rapidly to the entire body within a few days or even within a couple of hours. The initial skin lesions are characterized by erythematous, dusky-red, irregularly-shaped, purpuric macules, which coalesce progressively (Fig. 1). Atypical target lesions with dark centres are often observed. Confluence of necrotic lesions leads to extensive erythema, and Nikolsky’s sign (dislodgement of the epidermis by lateral pressure) is positive on erythematous areas. Flaccid blisters that burst easily are present at this stage, and the necrotic epidermis is easily detached at pressure points or by frictional trauma, revealing large areas of exposed, red, sometimes oozing dermis, whereas the epidermis may remain in other areas (10, 15, 18, 33).
In terms of severity, cases are classified according to the consensus definition (Table I) based on the total area in which the epidermis is detached or detachable (positive Nikolsky’s sign). Correct evaluation of the extent of detachment is difficult, especially in areas with spotty lesions and small blisters. Therefore, it may be helpful to remember that the surface area that can be covered by one hand (the patient’s hand in children) represents approximately 1% of the patient’s BSA (10, 15).
Mucous membrane involvement (in most cases on at least 2 sites) is observed in approximately 90% of patients (Fig. 2). It typically begins with erythema, followed by painful erosions of the oral, ocular, genital, nasal, anal and, sometimes, tracheal or bronchial mucosa. These symptoms usually lead to impaired alimentation, photophobia, conjunctivitis and painful urination. The oral cavity is almost invariably affected and reveals painful haemorrhagic erosions, often with greyish white pseudomembranes. The lips are covered with haemorrhagic crusts. Approximately 80% of patients have conjunctival lesions accompanied by pain, photophobia, lacrimation, redness and discharge. Severe forms may lead to epithelial defect and corneal ulceration, anterior uveitis, and purulent conjunctivitis and blepharitis. Synechiae often occur between eyelids and conjunctiva, and eyelashes may be shed. Genital erosions are frequent in men and women, but may be more easily overlooked in females, especially in young girls.
To detect such distinct features requires a thorough clinical examination of the patient’s entire body, involving further specialists in the examination of eyes, deep throat and genital mucosa in women. Ophthalmological consultation, in particular, is an urgent requirement to prevent complications and long-lasting sequelae (10, 15, 18, 33).
Although more than 100 different drugs have been reported in the literature as inducers of EN, less than a dozen have been identified to carry a high risk, and these account for more than half of the cases occurring in Europe according to 2 multinational case-control studies (16, 34). These high-risk drugs are allopurinol, antibacterial sulphonamides, certain antiepileptic drugs, such as carbamazepine, lamotrigine, phenobarbital and phenytoin, non-steroidal anti-inflammatory drugs (NSAIDs) of the oxicam-type, and nevirapine. The risk appears to be confined to the first 8 weeks of treatment and most reported EN cases started after the first continuous use of the medication between 4 and 28 days (16, 34, 35, 36). For lamotrigine and the anti-HIV-drug nevirapine, it was thought that a slow titration of the dosage could prevent such severe adverse reactions, since slow dose escalation had been shown to decrease the rate of mild eruptions. However, there is no evidence for a decreasing risk of EN (37–39). Oxcarbazepine, a 10-keto derivative of carbamazepine, which was considered to have a far lower risk, seems to cross-react with carbamazepine, revealing a lower, but substantial, risk of causing EN. Allopurinol, an old drug used to treat hyperuricaemia and gout, is widely believed to be a very safe medication; however, it was identified as the major cause of EN in Europe and Israel more than a decade ago and remains as such to date (40, 41).
Often the entire group of NSAIDs is suspected to induce EN, but there is a huge difference in risk among the various groups: oxicam derivatives carry the highest risk, acetic acid derivatives (e.g. diclofenac) moderate risk, and propionic acid derivatives (e.g. ibuprofen) no increased risk (Table III) (34, 36).
Table III. Drugs and recommendations in epidermal necrolysis (EN) (34)
Among anti-infective agents, a significant, but much lower, risk than for antibacterial sulphonamides has been shown for different groups of antibiotics, such as cephalosporins, quinolones, tetracyclines and aminopenicillins. For other medications, such as corticosteroids, proton pump inhibitors or tramadol, the calculated risk was strongly affected by confounding (16, 34). In comparison with the results of 2 case-control studies, recent analysis of systematically ascertained registry data on EN using ALDEN (algorithm for causality assessment in EN) (42) demonstrated that the proportion of validated cases that could be explained by medications with a significant (high and moderate) risk was stable (65–68%) over a period of more than 2 decades (16, 34, 42). ALDEN provides structured help for identifying the most likely culprit drug and is based on the following criteria: time latency between start of drug use and index-day (i.e. onset of the adverse reaction), drug present in the body before index-day (taking into account the drug’s half-life as well as the patient’s liver and kidney function), information on prechallenge/rechallenge and dechallenge (if available), type of drug/notoriety (based on drug lists that require a regular update) and alternative causes. Numerical score values lead to a causality assessment for each individual drug a patient has taken or was administered, ranging from “very unlikely”, “unlikely”, “possible”, “probable” to “very probable (43). For approximately one-third of cases of EN, no patent drug cause could be identified by using 2 completely different epidemiological methods. Even if new drugs or combinations of old drugs are taken into account as triggers of EN, at least 25% of all cases remain without a plausible drug cause, whereas this proportion reaches 50% among children and adolescents with EN. In these cases other eliciting factors must be saught:
An important non-drug risk factor is infections within one month before reaction onset. Most often these infections are diagnosed by clinical means, but positive serology related to certain well-known infectious agents, such as Epstein-Barr virus, cytomegalovirus, adenovirus or Mycoplasma pneumoniae, are rare. In some cases a preceding infection cannot be distinguished from the prodromal symptoms of EN; in others the reaction occurs suddenly with no prior signs or symptoms and must be labelled as “idiopathic” (10, 24).
EN has also been reported in the context of bone marrow transplantation, some eruptions of which may be induced by medication use, others are rather a maximal variant of acute graft-versus-host disease (GVHD). However, clinical and histological findings in EN and extensive acute GVHD are often indistinguishable, but depending on reaction onset after transplantation and the presence of non-cutaneous symptoms of GVHD, this diagnosis seems to be more likely (44). Lupus erythematosus (systemic LE or subacute cutaneous LE) is associated with an increased risk of EN. Often drug causality is doubtful in such cases and keratinocyte necrosis and subsequent skin detachment may be an extreme phenotype of cutaneous LE that must be considered as a differential diagnosis of EN (45).
For drug analysis in epidemiological studies, as well as for causality assessment in an individual case of EN, the correct determination of the day of reaction onset (so-called index-day) is of major importance (10, 15, 33, 34). All medications taken within a month preceding the index-day should be listed with their first and last day of use. Furthermore, information on prior use is very important, since it is rather unlikely for a medication to be the cause of EN if it was taken and tolerated in the past. A drug inducing EN is typically taken as the first continuous use, most often for 1–4 weeks, but sometimes for up to 8 weeks, without prior exposure (34). Thus, the mechanism differs from the classical sensitization in allergic conditions (10, 15, 37, 38, 39).
Frequently, and especially when no obvious drug cause is identifiable, medications taken to treat the prodromal symptoms are suspected of having induced the reaction. This mainly concerns antipyretics, analgesics, and secretolytics, sometimes summarized as “cough and cold medicines.” When looking more closely at the use of these medications, they have usually been taken and tolerated previously and/or were started after the onset of prodromal symptoms of EN (“protopathic bias”). Neither of these patterns is typical for drug exposure causing EN (15, 33, 46). In contrast, medications causing EN have not been used previously and their exposure represents the first continuous use that started 4 weeks to at least 4 days before reaction onset. Furthermore, these substances do not belong to the drug groups for which an increased risk was estimated in epidemiological studies (34, 35). Differentiation between infection and drugs as the triggering agent can be challenging in the case of antibiotics used to treat infections (“confounding by indication”), but it helps to consider the type of infection, since classic bacterial infections alone do not seem to have an increased risk of causing EN (33).
For GBFDE, there are numerous case reports in the literature providing information on possible drug triggers (47–50); however, no analyses have been conducted on large patient numbers. The range of triggers include antimicrobial sulphonamides (especially cotrimoxazole), analgesics (especially metamizole, but also paracetamol), and, less frequently, antibiotics, allopurinol, and antiepileptic drugs (especially carbamazepine) (47–50). The latency between the start of drug use and reaction onset ranges from a few hours to a few days. In contrast to EN, the triggering agent has often been used and tolerated in the past (18). Sensitization happens over time, meaning that a reaction consistent with a fixed drug eruption occurs rapidly upon renewed use of the drug. Thus, GBFDE is a classic allergic reaction that must be differentiated from EN.
The risk of recurrence in EN appears to be rather low, as Kirsti Kauppinen had already observed in 1972 (5). In the multinational RegiSCAR study, few individual patients experienced a second event of EN after accidental exposure to the same drug that had induced the first event. The time latency between the start of drug use and reaction onset was very similar and not necessarily shorter, as reported repeatedly in the literature.
In contrast, fixed drug eruption, including GBFDE, has a high risk of recurrence, which may be explained by memory T cells remaining in the affected skin (51). In many cases there has been a previous, often less severe, event, but cases with extensive skin detachment may also occur de novo and re-occur with the same amount of involvement (49).
EMM appears to be almost exclusively triggered by infections, especially M. pneumoniae in children and adolescents, and herpes simplex virus in adults. Recurrence has been observed, in up to 10% of cases, and in some patients even several times, before the reaction resolves (20). Interestingly, infection-induced EN cases do not seem to recur, and it may be assumed that the viral triggers change so rapidly that they are not recognized again as an antigen (52).
A T-cell reaction comparable to GVHD is believed to be the pathogenetic mechanism in EN, since immunohistochemical investigations identified primarily CD4+ cells in the dermis and CD8+ cells in the epidermis (53, 54). In contrast to what was postulated in earlier years, these cytotoxic T cells are usually specifically directed against the native form of the drug rather than against reactive metabolites (55). The acute necrosis of keratinocytes in EN is attributed to an extensive process of apoptosis (54, 56). Cytotoxic T-cells are able to initiate apoptosis, enhanced by the release of perforin and cytokines, such as TNF-α or granzyme B (57, 58). It is also assumed that proteins such as Fas antigen (CD 95) and the P55 TNF-α receptor enhance apoptosis in keratinocytes (59). However, it was demonstrated that Fas and Fas ligand are not the most important cytokines in the acute phase of EN, but rather the cationic protein granulysin (60). It showed the strongest cytotoxicity in the blister fluids of patients with EN compared with other blistering diseases, with its concentration correlating with the severity of the clinical reaction (60). Therefore, it was concluded that granulysin is a severity marker in EN and provides a target for possible immunomodulating treatments. It has also been shown that IL-15 is associated with the severity of the reaction as well as the risk of mortality (61).
It has been known for many years that there is a genetic predisposition to develop EN. As early as 1987, different human leukocyte antigen (HLA) loci were found for TEN associated with sulphonamides or with oxicam-NSAIDs (62). Almost 20 years later, a strong association between HLA-B*1502 and carbamazepine was observed in patients with EN who were of Han Chinese descent (63). This association could not be detected in European patients, where HLA-B*5701 was identified to confer genetic susceptibility to carbamazepine-induced SJS/TEN (64). Interestingly, HLA-B*5701 had previously been demonstrated to be associated with abacavir hypersensitivity, which is characterized by fever, rash and constitutional, gastrointestinal, and/or pulmonary symptoms different from SJS/TEN and DRESS (52). A second strong association with HLA-B*5801 was observed in Han Chinese patients with allopurinol-induced disease, not only for EN, but also for DRESS (65). For this allele an association of 55% was found in allopurinol-induced EN cases of European descent (66). Clearly, genetic predisposition is not the only important factor for developing a certain type of severe cutaneous adverse reaction due to a specific drug, but also the patient’s ethnicity, as was shown for patients of southeast Asian, European and African descent (52).
To date, there have been no systematic investigations into the genetic pattern of infection-induced EN cases. However, some reports on specific HLA alleles in cases thought to be triggered by antipyretics and secretolytics appear to be ultimately associated with infection-induced reactions (46). Although a large genome-wide association study in European patients with EN demonstrated that the relevant alleles/genetic variants are all located in the HLA locus on chromosome 6, the variability in the European population appears to be too large to deploy a medication-specific predictive test to prevent EN (67). In contrast, this has been successfully demonstrated in Southeast Asian subjects, at least in the case of carbamazepine, for which the predictive test has led to a marked reduction in carbamazepine-induced EN cases (68).
Although no systematic investigations into the pathogenesis of GBFDE have yet been undertaken, there are analyses on the T-cell population in fixed drug eruption. T cells play an important role here, since they remain in the affected areas of skin as “memory cells”, which explains why a reaction re-occurs at the same site. The term “fixed drug eruption” takes this fact into account, although the reaction may expand if it recurs (51). Furthermore, several cytokines, such as FAS/FAS-L, perforin and granzyme B, are equally expressed in GBFDE and EN, whereas the concentration of granulysin is much lower in GBFDE compared with EN (27).
Taking a detailed and thorough medication history is crucial. Assuming a medication rather than an infection triggered the reaction, the most likely culprit drug should be identified and discontinued. Thus, it is essential to know the time latency between the start of drug use and onset of the reaction, as well as the drugs that have a high-to-moderate risk of the type of reaction in question. It may be helpful to create a timeline diagram, into which the chronological sequence of clinical symptoms is entered on the x-axis and the medications taken or applied are entered on the y-axis (Fig. 5). Based on the diagram and the information on duration of use (start and end of use), it is possible to narrow down or even identify the inducing agent. It then becomes obvious that not all drugs, some of which may be vital for life, need to be withdrawn. Medications that were administered to treat prodromal symptoms and that are often suspected as the cause of EN, can also be excluded as triggers. If an infection is thought to have induced the reaction, patients should receive adequate antibiotic or antimicrobial treatment; reluctance to provide medication in a medical condition frequently caused by drugs may be detrimental (15, 33). The following supportive care and topical treatment is recommended:
In order to assess a patient’s prognosis and to decide on the appropriate therapeutic options, the SCORTEN (severity-of-illness score for EN) has been developed (69). Seven independent, but equally significant, factors are used for the calculation of score points: (i) age (≥ 40 years), (ii) heart rate (≥ 120/min), (iii) malignancy, (iv) percentage of detachment relative to BSA on day 1 (≥ 10 %), (v) serum urea (> 10 mmol/l), (vi) serum bicarbonate (< 20 mmol/l), and (vii) serum glucose (> 14 mmol/l) (69). The positive score points are added and the higher the value, the higher is the risk of death and the lower the chance of survival (69–71) (Table IV).
Fig. 5. Timeline diagram with chronologic sequence of clinical symptoms (x-axis) and medication use (y-axis).
Table IV. SCORTEN (69) (severity-of-illness score for epidermal necrolysis) to assess a patient’s prognosis
Only patients with limited skin involvement, and a SCORTEN value of 0 or 1, and a disease that is not rapidly progressing can be treated in non-specialized wards. Depending on the local or national facilities, patients who do not need intensive care may remain in dermatology units or hospitals (in many European countries), others should be transferred to intensive care facilities or burn units (72, 73). Supportive care is still the cornerstone of treatment and includes maintaining haemodynamic equilibrium and preventing life-threatening complications. Due to significant fluid loss in patients with large amount of skin detachment, hypovolaemia and electrolyte imbalance must be adjusted on a daily basis. Infusion volumes are usually lower than for burns of a similar extent of skin detachment (approximately 1/3–/4 of the infusion volume in burns) because interstitial oedema is absent. In order to select the correct amount of fluid replacement, correct estimation of the denuded BSA is important (74). Peripheral venous lines should be used, if possible, since the sites of insertion of central lines are far more prone to infection. Increasing the environmental temperature to 25–30°C is important to compensate for loss of thermoregulation in patients with extensive skin detachment (72). Air-fluidized beds may help to increase the patient’s comfort. To reduce the risk of infection, aseptic and careful handling is required. Skin, blood, and urine specimens should be cultured for bacteria and fungi at frequent intervals. Prophylactic use of antibiotics should be avoided, and instead patients with EN should receive antibiotics when an infection is suspected based on clinical features and laboratory results. Prophylactic anticoagulation is needed and early nutritional support should be provided through nasogastric tubes in order to promote healing and decrease the risk of bacterial translocation from the gastrointestinal tract (10, 18, 72, 73). For adequate enteral nutrition, intensive care guidelines (e.g. ESPEN guidelines) should be followed (75).
Topical treatment plays a special role in bullous reactions. Antiseptic solutions or gels, as well as non-medicated and non-adhesive gauze dressings are used. There is no standard policy concerning the use of antiseptics and wound dressings, which remains a matter of experience in each centre. Careful handling and skilful wound care, performed by experiences nurses, in addition to adequate pain management, are essential (10, 72, 73).
Some experts recommend leaving the blister roof in place as a natural cover to protect the dermis, while others recommend complete removal of detached skin and the consecutive use of biosynthetic dressings in order to protect against infection. Although this remains a topic of debate, it was recently suggested that aggressive debridement is neither necessary in superficial burns nor in EN, because superficial necrosis is not an obstacle to re-epithelialization and might even accelerate the proliferation of stem cells due to inflammatory cytokines (76).
In the case of erosive mucous membrane involvement, local antiseptic treatment is recommended and the appropriate medical specialist should be consulted. In terms of eye involvement, an experienced ophthalmologist should examine the patient immediately after admission. Preservative-free emollients, antibiotic or antiseptic eye drops, often alternating with anti-inflammatory (e.g. corticosteroid) eye drops are recommended every 2 h in the acute phase. In case of early synechiae, mechanical disruption is indicated and graft of cryopreserved amniotic membrane has been proposed to decrease the rate of severe ocular sequelae. In any case, severe ocular involvement requires daily consultation with an ophthalmologist (73, 77).
Disinfectant mouthwash can be used for treatment of oral erosions, whereas erosions of the lips should be treated with bland ointment, e.g. dexpanthenol. Genital erosions in male and female patients may lead to adhesions or strictures. To avoid such complications wet dressings or a sitz bath are helpful. If deeper vaginal involvement is suspected in young girls, a gynaecological examination should also be performed, since early adhesions must be carefully disrupted. To avoid these, dilators covered with ointment can be applied (78).
Since GBFDE is considered to be a self-limiting disease that ceases to progress shortly after discontinuation of the triggering drug, supportive care alone is adequate. However, complications requiring intensive care can occur, especially in older patients and patients with extensive skin detachment. Topical treatment is the same as in EN. Since the mucous membranes are most often unaffected, interdisciplinary consultations are not mandatory, but can be helpful in some cases (24, 48).
Immunomodulating treatment. Because of the immunological mechanisms with involvement of cytotoxic T-cells and release of cytokines, several immunosuppressive and anti-inflammatory treatments have been tried to halt the progression of the disease. Data on therapeutic approaches largely derive from uncontrolled case series and case reports. Due to the rarity of SJS/TEN and the resulting low patient numbers, as well as the unexpected onset and rapid progression of the reaction, it remains a huge challenge to conduct a controlled randomized study on treatment efficacy. Therefore, existing data on treatment of EN must be evaluated with care:
To date, there are no data from clinical trials on the benefit of systemic immunomodulating therapy in the treatment of GBFDE. Systemic glucocorticosteroids are also used in some patients, but it appears that their short-term use does no harm and does not result in faster healing (18, 49).
During the acute stage of the disease, EN may be accompanied by hepatitis, tubular nephritis, or tracheobronchial mucosal involvement, which usually resolve rather quickly (10, 73). The most common complications include nosocomial infection and septicaemia, frequently caused by central venous catheters. Therefore, peripheral catheters should be preferred wherever possible and specific hygiene measures are advised, e.g. reverse isolation, etc. (72, 73).
The majority of EN survivors experience long-term sequelae of varying severity, affecting primarily the skin and mucous membranes (106, 107). Whereas skin lesions generally heal without scarring, hyper- and hypo-pigmentation of the skin as a result of the inflammatory reaction often persist for months to years. Reversible loss of hair and nails, as well as nail growth disorders are frequently observed. Mucosal adhesions that may cause strictures in, for example, the urethra or oesophagus, represent a greater problem. By far the most hazardous and, for the patient, most dramatic, sequelae affect the eyes by symblepharon formation with entropium and trichiasis, which can even cause blindness (10, 15, 77, 106, 107).
Many patients still experience somatic as well as psychological sequelae years after their reaction. These sequelae may range from symptoms of post-traumatic stress, sleep disorders, and nightmares, to fear of using any medications. A large survey, performed 5 years after EN, revealed that many patients and their relatives are inadequately informed about their reaction, its sequelae, and how to deal with these in the long term (107).
EN is not an allergic reaction in the strict sense, since there is no classic sensitization as in other delayed hypersensitivity reactions. In the latter, initial use of the substance is well tolerated, with a reaction developing only upon renewed exposure. EN differs in that it typically occurs during the first course of treatment with a drug (34).
GBFDE, on the other hand, is a true allergic reaction, since previous exposure to the triggering drug has usually occurred, and repeated use often causes localized fixed drug eruptions. While renewed administration of a triggering drug in patients with GBFDE can be expected to cause a rapid onset, and possibly even more extensive, repeated reaction, EN was rarely observed following similar re-exposure (5).
Skin tests, such as the patch test, are generally safe, but most often are not helpful for confirming the suspected trigger in EN. The success of testing depends, to a great extent, on the type of reaction and the T-cell populations involved, as well as on the drug to be tested. In a study performed a few years ago in France, for example, the triggering agent was confirmed by patch testing in less than 25% of patients with EN (108). One should also bear in mind that allopurinol, a very common trigger of EN, is not suitable for skin testing due to the lack of lipophilicity and skin penetration (108, 109).
In vitro tests were the most suitable instrument to identify the inducing agent in bullous drug reactions; however, their use is yet not part of routine diagnostics and remains rather experimental. This may, in part, be due to the fact that the specificity of the various tests, e.g. the lymphocyte proliferation test, the lymphocyte stimulation test, and cytokine assays, is high, while their sensitivity is much lower (109).
The German Registry of Severe Skin Reactions (dZh), representing the German part of the multinational RegiSCAR-study since 2003, was mainly funded by a research grant from the European Commission (QLRT-2002-01738) and by a grant from the Federal Ministry for Education and Research (Bundesministerium für Bildung und Forschung (BMBF); grant no. 01KG1018). The dZh also received a grant / donation by Erika- and Werner Messmer-Foundation for clinical research (grant no. 1020.0355.01a), a private donation (C.H.R., Nailsea, UK) for SCAR-research (grant no. 1020.0355.01b) and a grant/donation by the German Dermatology Foundation (Deutsche Stiftung zur Förderung wissenschaftlicher Arbeit auf dem Gebiet der Dermatologie; grant no. 1020.0355.01c). A minor part of financial support was provided by several pharmaceutical companies (Bayer vital, Boehringer-Ingelheim, Cephalon, GlaxoSmithKline, Grünenthal, MSD Sharp and Dome, Merck, Novartis, Pfizer, Sanofi-Aventis, Servier, Tibotec-Janssen) between 2003 and 2012. The author received the Else Kröner Memorial Stipendium for support of clinical research through Else Kröner-Fresenius-Foundation. Methodological considerations were partly supported by German Research Foundation (Deutsche Forschungsgemeinschaft; FOR 534).


 Click to show fullsize
Click to show fullsize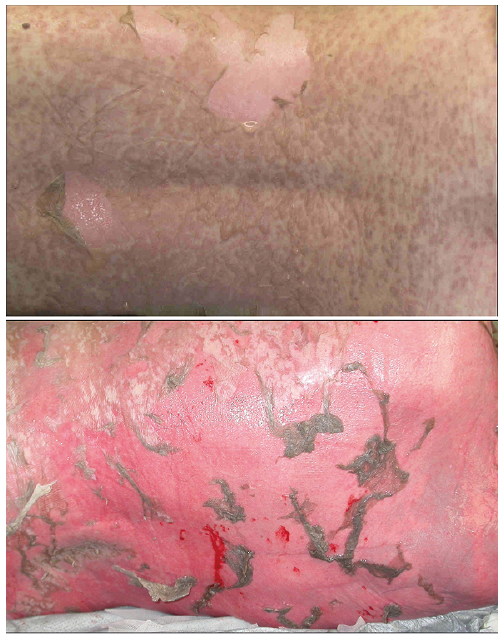 Click to show fullsize
Click to show fullsize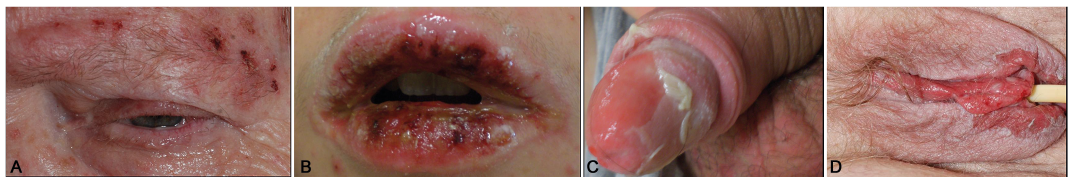 Click to show fullsize
Click to show fullsize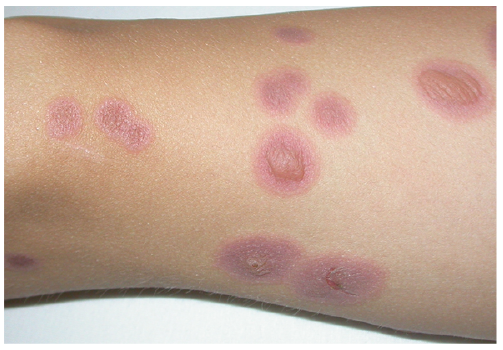 Click to show fullsize
Click to show fullsize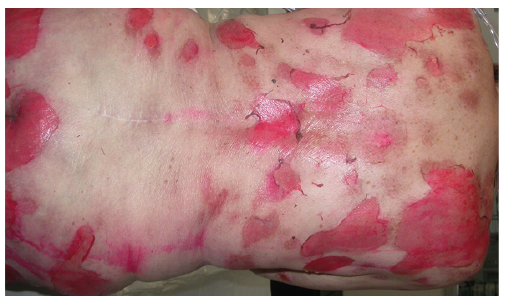 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize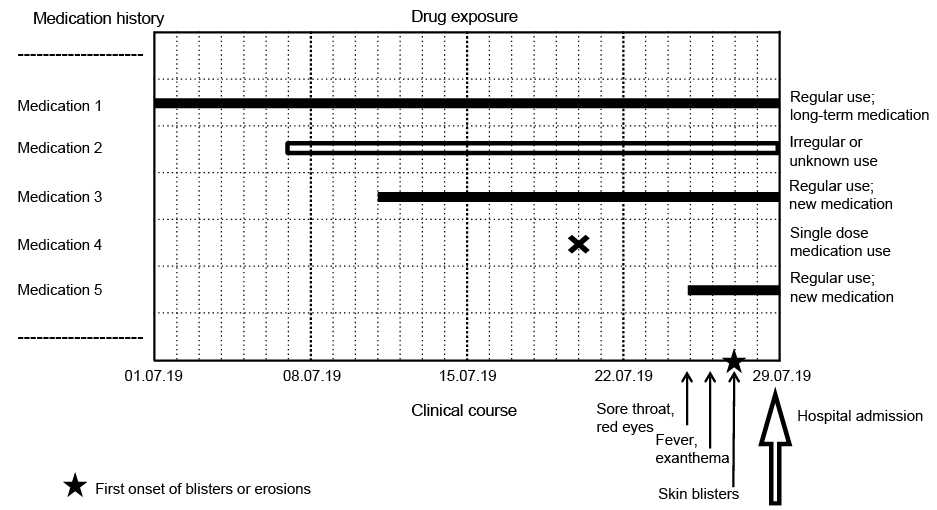 Click to show fullsize
Click to show fullsize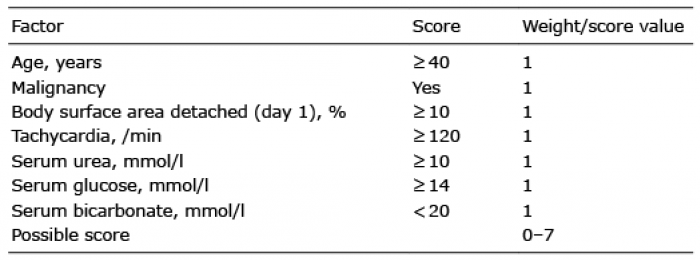 Click to show fullsize
Click to show fullsize