


1Curie Institute, PSL Research University, INSERM U1021, Normal and Pathological Development of Melanocytes, 2CNRS UMR 3347, University Paris-Sud, University Paris-Saclay, Orsay, France, 3Equipe Labellisée Ligue Contre le Cancer, 4Department of Biochemistry and Molecular Biology, Biomedical Center, Faculty of Medicine, University of Iceland, Reykjavik, Iceland, 5Laboratory of Biological Pharmacology, St Louis Hospital, Paris, France, and 6Department of Biopathology, INSERM U1186, Gustave Roussy, University Paris-Saclay, Villejuif, France
Cutaneous melanoma arises from melanocytes following genetic, epigenetic and allogenetic (i.e. other than epi/genetic) modifications. An estimated 10% of cutaneous melanoma cases are due to inherited variants or de novo mutations in approximately 20 genes, found using linkage, next-generation sequencing and association studies. Based on these studies, 3 classes of predisposing melanoma genes have been defined based on the frequency of the variants in the general population and lifetime risk of developing a melanoma: (i) ultra-rare variants with a high risk, (ii) rare with a moderate risk, and (iii) frequent variants with a low risk. Most of the proteins encoded by these genes have been shown to be involved in melanoma initiation, including proliferation and senescence bypass. This paper reviews the role(s) of these genes in the transformation of melanocytes into melanoma. It also describes their function in the establishment and renewal of melanocytes and the biology of pigment cells, if known.
Key words: melanocyte stem cells; embryonic development; germline mutation; inherited melanoma.
Accepted Apr 27, 2020; Epub ahead of print Apr 28, 2020
Acta Derm Venereol 2020; 100: adv00139.
Corr: Lionel Larue, Institut Curie, PSL Research University, INSERM U1021, Normal and Pathological Development of Melanocytes, Orsay, France. E-mail: lionel.larue@curie.fr
Inherited variants or de novo mutations in approximately 20 genes have been shown to contribute to approximately 10% of cases of cutaneous melanoma. This paper evaluates the function(s) of these proteins in the establishment of the lineage during embryogenesis, melanogenesis, renewal and, of course, during melanomagenesis.
Cutaneous melanoma results from transformation of melanocytes (Mcs). Melanoma accounts for only 10% of skin cancers, but is responsible for approximately 80% of skin cancer-related deaths; the remaining skin cancers are basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and Merkel cell carcinomas. The incidence of melanoma has increased steadily over the last 5 decades. According to Berk-Krauss et al. (1), the overall mortality from 2013-2016 in the US among caucasians was 6%. Melanoma accounts for approximately 2% of all cancers diagnosed annually worldwide (2, 3). Phototype (Table I) and geographical location are two key risk factors in the epidemiology of cutaneous melanoma: melanoma incidence is highest in Australia/New Zealand, followed by the USA and Northern Europe, and mostly affects Caucasian populations of phototypes I and II (4). It has been shown that for individuals living in similar environments in the USA, Caucasians have a 25 times higher risk of developing melanoma than African Americans (phototypes IV to VI). The risk of melanoma in red-haired individuals is approximately 3 times that observed in other Caucasians (5). Caucasians were found to have a 4–5 times higher risk of developing and dying from melanoma in Australia than in Europe, showing the effect of the environment. The incidence of melanoma is 56 per 100,000 in Australia and 14 in 100,000 in France, with a similar mortality (approximately 1/100,000).
Table I. Phototypes: Fitzpatrick classification
The frequency of melanoma is lower in dark-skinned individuals than in light-skinned individuals. This may be explained in part by the melanoma inducers present in the environment (ultraviolet radiation). Epidemiological studies have revealed other melanoma inducers, including heavy metals, some pesticides, and alcohol consumption, but none of these were associated with phototypes (6–9). These fundamental data reveal the importance of genetics (mainly senescence, pigmentation and DNA repair genes) and the environment (mainly sun/ultraviolet (UV) exposure) in melanomagenesis.
Melanomagenesis is a multistep process that can be divided into 2 main stages: initiation and progression. Melanoma initiation requires proliferation of Mcs and bypassing of senescence. After a certain number of divisions, cells enter senescence and, in order to bypass senescence and allow the further growth of the cells, the cell cycle and the length of the telomeres have to be boosted.
Mcs are melanin-producing cells that arise from neural crest cells (NCC) (sometimes called the 4th embryonic layer), a transient population of cells arising from the dorsal part of the neural tube (10). Information concerning the establishment of the Mcs was largely obtained from mouse and chicken studies. The NCC delaminates and migrates away from the neural tube. NCC derivatives exist as single cells throughout development and spread via 2 major migration pathways. Melanoblasts are NCC derivatives migrating in the space between the somites and the non-neural ectoderm (dorsolateral pathway). These cells are the precursors of Mcs and are characterized by an ability to produce the pigment melanin. They are specified in a cell-free area between the dorsal part of the neural tube, the ectoderm and the dorsal part of the somites. This area is known as the migration staging area (MSA), the site at which founder melanoblasts receive proliferation, survival and migratory signals (11, 12). Melanoblasts arising from the dorsolateral pathway travel through the developing dermis. From mid-gestation onwards, the dermal melanoblasts start to cross the basal layer and colonize the epidermis (13). The epidermal melanoblasts then concentrate around the placodes of hair follicles (HF) before entering the forming hair follicles (13, 14). They colonize the future bulge, to generate the melanocyte stem cells (McSC), and the hair bulb, to generate the differentiated Mcs of the first hair cycle (“embryonic” hair), before resting. In addition to colonizing the hair follicle in humans and pigs, melanoblasts also remain in the epidermis in the interfollicular regions. Mcs located in the interfollicular regions are responsible for the tanning response and the protection against UV. Furry animals do not require such protection, since the hair efficiently protects the skin against this type of radiation.
Hair renewal and pigmentation are concomitant processes. Mcs disappear during catagen. In early anagen, McSCs re-enter the cell cycle and divide, for self-renewal and the generation of transit amplifying cells (TAC). These cells migrate and differentiate into Mcs, which participate in the pigmentation of the first “adult” hair. Cutaneous melanoma may arise from the McSCs, TACs and/or from the Mcs (15).
Approximately 20 genes have been found to be constitutively mutated in the germline and associated with a risk of melanoma. Three classes of genes have been defined on the basis of the frequency of the variation and the risk of developing melanoma: ultra-rare variants with a high risk, rare variants with a moderate risk, and frequent variants with a low risk (Fig. 1). It has been estimated that ultra-rare variants conferring high risk (~10 genes) account altogether for 2% of the total risk of melanoma. These genes include p16INK4A and p14ARF (located at the same locus, CDKN2A), CDK4, BAP1, RAD51B, POLE, TERT, POT1, ACD, and TERF2IP. Rare variants of MC1R and MITF confer a moderate risk of melanoma and finally, frequent variants of melanocortin 1 receptor (MC1R), OCA2, ASIP, TYR, TYRP1, MATP, SCLC45A2, KIT, and PARP1 are estimated to account for 12% of the risk of melanoma. It should be noted that the melanoma risk model is very complex, as genetic risk factors interacts together (rare and frequent variants modulate the risk conferred by ultra-rare variants (16)), but also with host phenotypes and environmental factors (17). Functional studies performed in vitro and/or in vivo has unravelled the role of some of these genes in melanoma. However, no systematic study has yet been performed on all 20 genes in order to evaluate their importance during the natural course of Mcs development and melanomagenesis.
This review focusses first on susceptibility genes for cutaneous melanoma and the melanoma inducers found in the environment. It then discusses the role of these genes, if known, during the various steps of Mcs and melanoma development, including in: (i) melanoma initiation and progression, (ii) the establishment of the Mcs lineage during embryonic development, (iii) the terminal differentiation of Mcs associated with the production and transport of melanin, and (iv) the transfer of melanosomes in the keratinocytes of the skin or the hair, (v) the renewal of Mcs from McSC, and (vi) the maintenance of Mcs function over time.
Fig. 1. Interaction network of genes involved in melanocyte biology, and the associated melanoma risk. The list of genes involved either in the establishment and renewal of melanocytes, in the biology of pigment cells, and/or associated with increased melanoma risk, was submitted to the STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) database for analysis of the gene interaction network (122). Each circle represents 1 gene, and 1 linking line represents a direct (physical) or indirect (functional) association between the proteins encoded by the 2 genes. Green: genes with ultra-rare variants associated with a high risk of melanoma; blue: genes with rare variants associated with a moderate risk of melanoma; red: genes with frequent variants associated with a low risk of melanoma; white: genes involved in melanocyte biology with no mutation currently associated with melanoma. Note, the left cluster gathers mainly genes of melanogenesis, and the right cluster gathers genes of the cell cycle, telomere length control, and DNA repair.
The focus here is primarily on germline/constitutive mutations increasing the risk of melanoma formation and the environmental factors modulating these risks by inducing somatic mutations, epigenetic and allogenetic modifications and/or modifying the micro-environment.
Melanoma susceptibility genes
Variants in melanoma susceptibility genes have been classified according to their frequency and the degree of risk. These variants are generally transmitted from germ cells, but the role of neo-mutations, micro-chimerism and somatic mutations should not be underestimated.
Ultra-rare variants – high risk of developing melanoma. The first genetic studies of familial melanoma mapped markers associated with melanoma risk to chromosome 9, and the 9p21 region in particular (18); subsequently, causal variants at the CDKN2A locus were identified (19, 20). The CDKN2A (cyclin-dependent kinase inhibitor 2A) gene encodes 2 proteins, p16 and p14, which inhibit cyclin-dependent kinase (CDK), thereby regulating cell cycle checkpoints. CDKN2A variants have been found in 10–40% of familial melanoma cases, depending on geographical location, but their prevalence is very low in the general population, either never described or with a minor allele frequency (MAF) < 0.001% (21, 22). CDKN2A variants confer a high risk of melanoma development with age- and geographical area-dependent variations: in melanoma-prone families ascertained through cancer clinics, the penetrance (a mean age-specific cumulative risk) at age 80 years was 58% in Europe, 76% in the USA and 91% in Australia (23, 24). However, at the same age penetrance was lower (28%) in population-based studies (25). Differences in melanoma risk between CDKN2A mutation carriers can be explained partly by the underlying pigment genes these individuals also carry and possibly also sun-exposure variants, host phenotype and sun exposure (17, 26, 27).
Mutations of CDK4 (cyclin-dependent kinase 4), encoding another cell cycle regulator, have also been linked to melanoma formation in families with phenotypes similar to those of families with CDKN2A mutations (28, 29). CDK4 is a kinase that regulates G1 cell cycle progression by phosphorylating Rb proteins. CDK4 variants are rare and are found in less than 1% of familial cases of melanoma, with a penetrance of 74% at the age of 50 years (29).
More recently, high throughput sequencing studies have added to the list of genes increasing the risk of melanoma, through the identification of rare variants in families. These genes can be divided into 2 groups: TERT, POT1, ACD and TERF2IP, which encode proteins involved in telomere length control, and BAP1, RAD51B and POLE, which encode DNA repair proteins. A variant in the TERT (telomerase reverse transcriptase) promoter was identified in 2 melanoma-prone families; this
c.-57T>G variant is oncogenic through the creation of a new ETS transcription factor binding site leading to increased TERT expression (30, 31). Interestingly, somatic mutations of the TERT promoter (1 being identical) have also been detected in 33% of primary melanoma cases and 85% of metastatic cases (30). Mutations of POT1 (protection of telomeres 1), ACD and TERF2IP were detected in exome sequencing studies of melanoma-prone families (32, 33). These 3 genes encode proteins of the shelterin complex and explain 9% of melanoma families lacking CDKN2A and CDK4 mutations. Mutations of the TERT promoter and the shelterin complex result in longer telomeres, favouring senescence bypass.
BAP1 (BRCA1-associated protein-1) loss of function germline mutations have been found associated to a tumour predisposition syndrome (BAP1-TPDS, OMIM 614327) including both cutaneous (0.52% of families) and uveal melanomas (CM and UM) (28.5% of families) (34–37). In addition to melanomas, the most frequent cancers of this syndrome are renal cell carcinoma, mesothelioma and multiple BCCs but the whole tumour spectrum and lifetime risk are currently unknown (38). BAP1 is a tumour suppressor gene located at 3p21, encoding a deubiquitylase that participates in multi-protein complexes playing roles in numerous cellular processes, including DNA damage response, cell cycle regulation, cell growth, metabolism, and the regulation of inflammatory responses (38). The BAP1 loss-of-function mutations are associated with approximately 15% risk of cutaneous melanoma. A germline POLE missense mutation located in the exonuclease domain of the protein (p.(Trp347Cys)) was found in a unique melanoma-prone family of 7 confirmed cases of CM and a case with UM, leading to a mutator phenotype in functional assays (39). It should be noted that POLE germline mutations are more frequent in endometrial (7–10%) and colorectal (2%) cancers (22) than in melanoma. Finally, a novel germline RAD51B nonsense mutation was identified in a 3-case CM family; a melanoma tissue from a carrier displayed loss of RAD51B staining in most tumoural cells, by immunohistochemistry (40). RAD51B plays an important role in DNA repair through homologous recombination, but up-to-date known germline mutations have been associated with increased risk of ovarian cancer (41).
Approximately 22% of melanoma-prone families are associated with mutations in the 9 high-risk melanoma susceptibility genes (19% for CDKN2A and 3% for the other 8 genes). However, the remaining 78% can be partly accounted for by the rare variants conferring a moderate risk and frequent variants conferring a low risk of melanoma (24).
Rare variants – moderate risk of developing melanoma. A unique missense variant in the microphthalmia-associated transcription factor (MITF), the MITFE318K variant, was linked to melanoma risk in 2 different studies. One of the studies started as a candidate gene hypothesis for melanoma and renal cancer predisposition (42), whereas the other involved whole-genome sequencing of a melanoma-prone family (43). The assumption in the first study was that MITF might be a good candidate for the following reasons: (i) it has been proposed to act as a melanoma oncogene (44); (ii) it also stimulates the transcription of hypoxia inducible factor (HIF1A), the pathway of which is targeted by kidney cancer susceptibility genes (45); and (iii) two members of the MITF-family of proteins, TFE3 and TFEB, have been implicated in renal cell carcinoma through somatic translocations (46). Both studies identified the same rare MITFE318K variant (actual Minor Allele Frequency – MAF – in European population of 0.25%, GnomAD database) associated with a 5-fold increased risk of melanoma in genetically-enriched cases and an increased risk of 2.2 in case-controls studies (Yokoyama, 2011 #3370; Bertolotto, 2011 #2771).
The involvement of MC1R variants in melanoma is complex, since it is involved in pigmentation/phototype, naevi, UV/sun exposure (Demenais, 2010 #3357) and more recently, sex-dependence (47–49). The MC1R gene (16q24) is a key regulator of the synthesis of melanins (eu- and pheo-melanin) in Mcs, upon UV stimulation. Melanins are transferred to up to 40 surrounding keratinocytes to act as a natural sunscreen to absorb UV irradiation. Eumelanins are prevalent in phototypes IV to VI, while pheomelanins are responsible for red hair and freckles found in phototypes I and II (50). Beyond affecting phototypes, this 7-transmembrane receptor plays a role in DNA repair pathways and antioxidant defences for a complex photoprotective response (51). MC1R is highly polymorphic (> 80 variants) in Caucasian populations. Loss-of-function variants, which result in less pigmented phenotypes, are the result of human evolution associated with the classical mutation-selection events. Indeed Homo sapiens and Homo neanderthalensis migrated, and adapted from Africa to more northern and less sunny regions and subsequently lost pigmentation to allow increased vitamin D production (51). Six loss-of-function MC1R variants (p.ins86-87A, p.D84E, p.R142H, p.R151C, p.R160W, and p.D294H) are defined as R-type variants, as they increase the relative risk of melanoma 2–3 times; whereas another 4 variants (p.V60L, p.V92M, I155T, and p.R163Q) are classified as r-type variants as they confer melanoma risk below 2 (48). The effect is additive, as the presence of 2 or more MC1R “R” variants is associated with 6-fold increased risk compared to wild-type alleles. Thus, the MC1R gene is a moderate-penetrance melanoma susceptibility gene, but it is also a gene that modifies the risk of melanoma in patients with a CDKN2A mutation. Recent studies have shown that the risk conferred by MC1R variants seems to be independent of sun/UV exposure (52, 53) and independent of phenotype (54–56). Furthermore, MC1R polymorphisms seem to influence size and dermoscopic features of naevi (57). And recently, several sex-differences emerged, with MC1R variants associated with phototype I and II and higher melanoma risk, but better survival in females than in males (47–49).
Frequent variants – low risk of developing melanoma. Finally, frequent variants of genes associated with pigmentation (OCA2, ASIP, TYR [OCA1], TYRP1 [OCA3], MATP, SCLC45A2 [OCA4], KIT, and PARP1) are associated with a slight increase in risk of melanoma formation, as shown through genome wide case-control association studies (GWAS) (24, 58–60). These frequent variants have only a small individual effect on melanoma risk, but combinations of these low-risk variants may account for up to 78% of non-familial melanomas. They are also responsible for the host phototype and are therefore important for determining the interaction between the phenotype and the environment. Further functional studies are required to decipher the associated mechanisms. These frequent pigmentation variants probably play a role as modifiers of melanoma risk in other genetic backgrounds, such as the ultra-rare and rare variants, conferring high and moderate melanoma risk, respectively.
Environmental factors
Sequencing studies on tumours have highlighted the genetic complexity of melanoma in terms of the somatic mutational load in the population and melanoma is the cancer with the highest mean mutation rate (61). The spectrum of driver mutations provided unequivocal genomic evidence for a direct mutagenic role of UV light in melanomagenesis (62). Somatic mutation frequencies also differ considerably between melanoma patients, showing melanoma to be a complex disease with several subtypes and multifactorial origins (63).
The melanoma risk associated with the gene variants, described above, depends heavily on the co-occurrence of other gene variants and environmental (micro- [inside the body] and macro- [outside of the body]) stresses. Protein-protein, protein-microenvironment, and gene/protein-environment interactions undoubtedly account for the complexity and diversity of melanoma. Epidemiological studies have implicated several environmental factors in the induction of cutaneous melanoma, including ultraviolet radiation (UVR), alcohol use, obesity, heavy metals and some pesticides.
UVR from the solar spectrum is the leading external cause of melanoma formation. Epidemiological studies have shown that exposure to UV is correlated with melanoma formation: intermittent rather than chronic exposure, high levels of exposure during childhood and the use of artificial UV lamps are associated with a major risk of melanoma (64, 65). Both UVA (315–400 nm) and UVB (280–315 nm) can promote melanoma formation (66). Most melanomas have many somatic mutations and most of those have a UV signature (61), i.e. ≥ 60% of mutations are C→T at a dipyrimidine site, with ≥ 5% as CC→TT changes (67).
As already mentioned, several external factors seem to be associated with melanoma, including alcohol consumption, heavy metals and pesticide exposure (6–9), but the evidence for the associations obtained for alcohol consumption, heavy metals and pesticide exposure is weaker than that obtained for UV. Epidemiological studies on farm workers, a population also exposed to UVR, have highlighted a potential risk of pesticides. The cumulative effects of UV and the pesticide carbaryl are genotoxic in human Mcs (68). Obesity is associated with melanoma initiation and progression, as it increases the risk of melanoma formation and favours melanoma growth, invasion and distant metastasis (69). Indeed, adipose tissue favours the proliferation and aggressiveness of melanoma cells through a direct dialogue, mediated by soluble factors and by exosomes, and through remodelling of the tumour microenvironment. Little is currently known about the molecular mechanisms of melanomagenesis associated with alcohol, heavy metals and pesticides. Extensive in vitro and in vivo functional studies should be performed to validate the importance of these factors in melanomagenesis.
Melanoma susceptibility genes and environmental factors
One epidemiological study has revealed that CDKN2A mutation carriers appear to have the same cumulative risk of melanoma irrespective of the ambient UV irradiation of the region in which they live (27). These results were functionally validated after exposing mice lacking p16INK4A to transient UV irradiations; this did not affect melanoma formation (70). Of course, this does not mean that UV irradiation has no role in melanoma initiation, as there is a strong association between UV irradiation exposure and melanoma risk for the general population. Interestingly, UVB irradiation of mice expressing CDK4R24C, the oncogenic form of CDK4, promotes melanoma initiation, with a shorter time lag to tumour formation and faster growth (71). A germline nonsense mutation in BAP1 (Y646X) and environmental exposure to asbestos and UV irradiation were found to contribute to the high incidence of cutaneous melanoma in a family at high risk of cancer (72). The association of UV and MC1R mutations has long been known to promote melanoma, and MC1R has been shown to be a potent regulator of PTEN after UV exposure; interestingly, the major Red Hair Color (RHC) MC1R variants R151C, R160W and D294H where shown to bind PTEN less effectively than the wild-type protein (73).
Carcinogenesis is a multistep process. The wild-type cell accumulates genetic, epigenetic and allogenetic (it may affect transiently, for instance, RNA, proteins and lipids; which are modulated by the micro- and macro-environment) modifications, which alter its characteristics and/or environment, leading to self-sufficiency in growth signals, a limitless potential for replication, insensitivity to anti-growth signals, the evasion of apoptosis, sustained angiogenesis and tissue invasion and metastasis (74). A model of the multistep process of melanomagenesis has been described, in which the level of complexity is reduced to provide a schematic view of the process (75). Melanomagenesis is currently seen as a multistep process with 2 main stages: initiation and progression.
Initiation
Melanoma initiation is characterized by an initial “boost” of cell proliferation and the bypassing of senescence (75). It has been suggested that 25% of melanomas arise from naevi, 1–2-mm wide pigmented spots consisting of Mcs that have hyperproliferated in situ, but then stop proliferating and become senescent/quiescent. In the vast majority of naevi, the Mcs remain senescent throughout the life of the individual with a strict control provided by high expression of P14, P15, P16 and/or PTEN. However, in a few cases, melanomas arise due to a subsequent bypass of senescence. The remaining 75% of melanomas do not arise from naevi (76) suggesting that the initial proliferation of these cells is not affected by senescence. These 2 paths may appear different, but the molecular mechanisms are not; molecular mechanisms associated with the bypass of senescence may occur before those associated with proliferation. The RAS/MAPK signalling pathway is activated and involved in the proliferation step of most melanomas (77). Cell cycle proteins, such as CDKN2A/B and CDK4/6, and those of the PI3K/AKT and WNT/β-catenin signalling pathways are involved in senescence bypass/lack of senescence. PTEN loss and β-catenin activation can induce bypass of senescence or lack of senescence (78–81). Melanoma susceptibility genes are clearly involved in melanoma initiation.
Rare variants associated with a high risk of melanoma are involved in melanoma initiation. CDKN2A is certainly the best known of these genes. It encodes 2 proteins, one of which is p16INK4A, a CDK4/6 inhibitor that represses G1/S cell cycle transition, and is known to promote senescence. Its inactivation induces cell cycle progression and the bypass of senescence. It is therefore considered to be a tumour suppressor. Senescence is controlled by the cell cycle and by telomere length. TERT, POT1, ACD and TERD2IP, which regulate telomere length, may also be tumour suppressors because their inactivation induces checkpoint bypass and promotes uncontrolled cell cycle progression.
Rare variants associated with a moderate risk of melanoma development are also involved in melanoma initiation. Indeed, a rare variant of MITF (MITFE318K) has been shown to act through senescence bypass, leading to melanoma formation (42, 82). MITF is the main transcription factor of the Mcs lineage, where it regulates various functions, including melanogenesis/differentiation, proliferation, invasion, and senescence (83). MC1R variants modulate the incidence of melanoma, but it remains unclear whether this is linked to the protective effect of eumelanin against UVR or to the intrinsic role of MC1R in melanomagenesis. Studies to resolve this question are underway. One study revealed that MC1R mouse mutants in the BRAFV600E background develop more melanomas than control mice, independent of UV exposure. This highlighted the potential role of pheomelanin in inducing oxidative damage (52).
Frequent variants associated with a low risk of melanoma development are also involved in melanoma initiation. Melanoma incidence varies between populations; the higher the phototype the lower is the chance of cutaneous melanoma. Melanin, one of the key parameters for evaluating the level of the phototype, is a natural protector of the skin against external aggression. Variants in genes encoding proteins involved in melanogenesis are responsible for various forms of occulocutaneous albinism (OCA). These genes include TYR (OCA1), OCA2 (P), TYRP1 (OCA3), and SLC45A2 (OCA4). Recently, OCA5, OCA6 and OCA7 were identified and shown to correspond to SLC24A5 (OCA6), C10orf11 (OCA7), and a locus on chromosome 4q24 for OCA5. None of these have yet been associated with melanoma. However, further studies are required to fully evaluate the importance of these OCA genes in melanomagenesis. OCA1 variants have a general prevalence of 1/40,000, whereas OCA2 variants are more common in dark-skinned populations (Africa) (1/4,000 – 10,000) than in light-skinned populations (Caucasian) (1/36,000). OCA1 and OCA2 variants account for 80% of OCA cases and are the most strongly associated with skin cancer development. Melanomas and BCCs are more frequent in individuals with OCA1-2 mutations than in the general population. However, SCC is the most frequent type of cancer in patients with OCA mutations (84). Melanoma diagnosis is particularly challenging in this population because lesions are often amelanotic. Three other genes (ASIP, KIT, and PARP1) have frequent variants associated with a low risk of developing melanoma. Mutations of the KIT gene affect the tyrosine kinase receptor of the corresponding protein and cause piebaldism, as do mutations of the gene encoding its ligand, Steel (KITLG). KIT gene mutations are present in 39% of mucosal melanomas, 36% of acral lentiginous melanomas, and 28% of skin displaying chronic solar damage. Most of the reported mutations are found in exons 9, 11, 13, and 17, and they account for between 5% and 10% of the mutations of diagnosed melanomas (85, 86).
Progression
Tumour progression is characterized by the dissemination of the transformed cells, followed by the formation of metastases. Dissemination involves several fundamental cellular events, including a second boost of proliferation, pseudo-epithelial-to-mesenchymal transition, migration, intravasation of the blood and lymphatic streams, resistance to anoikis, and extravasation to invade new tissues. Cells may form metastases in a suitable niche, in which the cells induce angiogenesis and proliferate.
The function of MITF in melanoma progression is complex and can be explained with a rheostat model where the level and/or activity determine whether the Mcs or melanoma cells undergo senescence, invasion, proliferation or differentiation (83, 87, 87). MITF amplification is observed in 21% of metastatic melanomas and favours melanoma cell proliferation (44). However, MITF also represses proliferation through the regulation of p21 and p16 (88, 90). The functions of the proteins encoded by the other susceptibility genes in melanoma progression remain unknown.
We will now focus on the role of melanoma susceptibility genes in the establishment and maintenance of the Mcs lineage and pigmentation. The main function of Mcs, pigmentation, involves the intrinsic synthesis of melanin in specialized organelles called melanosomes, which are transported in Mcs and transferred to differentiating keratinocytes. Normal pigmentation is dependent on the genes involved in melanogenesis, and the transport and transfer of melanosomes and is finely regulated by extrinsic signals and cell-cell interactions.
Melanogenesis
Melanogenesis is a chain of reactions occurring in melanosomes, with tyrosine as an initial substrate, and pheomelanin (yellow, orange) and eumelanin (black, brown) as final products. The first enzyme in this chain, tyrosinase (TYR), hydroxylates tyrosine to generate DOPA, and then DOPAquinone. The second enzyme, dopachrome tautomerase (DCT or TRP2), and the third enzyme, tyrosinase-related protein 1 (TYRP1), catalyse eumelanin production from DOPAquinone. Pheomelanin production from DOPAquinone is dependent on cysteine.
Melanogenesis is regulated through modulation of the level, activity and localization of these enzymes by external signals, including communication between Mcs, keratinocytes and dermal fibroblasts via secreted factors and cell-cell contact. Mcs homeostasis is controlled by a complex network of keratinocyte-derived factors that regulate Mcs proliferation and differentiation. These include melanocyte-stimulating hormone (α-MSH), endothelins (Edn), basic fibroblast growth factor (β-FGF), nerve growth factors (NGF), granulocyte-macrophage colony-stimulating factor (GM-CSF), steel factor (SCF), leukemia inhibitory factor (LIF), hepatocyte growth factor (HGF), transforming growth factor beta (TGFβ), and Jagged1/2 (91, 92). These signals can be regulated by external factors, such as ultraviolet (UV) radiation, chemical compounds, drugs and stress.
The differences in pigmentation depend on the amount and quality of melanin (eumelanin vs. pheomelanin), which are partly controlled by the activity of MC1R. The MC1R receptor, which is activated by α-MSH after UV exposure, for example, induces eumelanin synthesis (93). MC1R variants are frequent in the Caucasian population, and lead to the expression of a receptor with normal, weak or no activity, associated with a brown, blond or red hair phenotype, respectively (94).
Melanosome transport
Melanosomes are lysosome-related organelles derived from non-pigmented endosomal vesicles (known as pre-melanosome stages I and II). They mature and undergo progressive pigmentation following melanogenesis and go through stages known as stages III and IV (95).
The mature melanosomes are transported from the perinuclear area toward the periphery of the Mcs and the tips of its dendrites. Two types of movement have been observed: rapid microtubule-directed migration over long distances, and short-distance migration along actin fibres at the periphery (96). During their migration from the nucleus toward the periphery of the cell (known as centrifugal movement), melanosomes are transported by a kinesin complex on microtubules. For the reverse migration toward the nucleus (known as centripetal movement), the melanosomes are transported by a dynein complex on microtubules. The motor for peripheral migration is myosin Va (dilute) in a complex with melanophilin (leaden) and RAB27A (ashen), and this migration takes place along actin filaments (97).
Mutations in genes encoding these transporters are associated with abnormal pigmentation, and, in some cases, more severe syndromes, such as Griscelli syndrome type 2 (98). However, they have not, as yet, been linked to an increase in melanoma risk. Conversely, the known melanoma susceptibility genes have not been shown to participate in melanosome transport.
Melanosome transfer
Melanosomes are transferred from Mcs to keratinocytes in order to deliver pigment to all epidermal keratinocytes. Several mechanisms of melanin transfer have been observed and debated: exocytosis-mediated, cytophagocytosis-mediated, tunneling nanotube-mediated and membrane vesicle-mediated transfer (99). The molecular mechanisms associated with the transfer of melanosomes to keratinocytes remain unclear. However, it has been suggested that classical pathways of exo-/endocytosis, membrane blebbing and vesicle biogenesis, filopodium formation and phagocytosis are involved. These processes involve proteins, such as Rab11b, small Rho GTPases, Cdc42 and Par-2 (100–103).
The cell body of the Mcs is located on the basement membrane, but the dendrites of the cell are in contact with 30–40 keratinocytes in the 3 dimensions of the epidermis, and these cells together form an epidermal melanin unit (104). In the basal layer, adjacent Mcs are generally separated by approximately 6 keratinocytes (105). Melanosomes containing melanin are located in the superior part of the keratinocytes protecting the nucleus against UVR.
Albino individuals, who have Mcs but lack melanin, rarely develop melanomas. However, they develop more carcinomas (BCC and SCC) than individuals with normal pigmentation, confirming the protective effect of melanin in keratinocytes. Several issues should be raised at this point. Albinism is associated with a number of vision defects, including photophobia. These individuals therefore tend to prefer to stay out of the sun, thus leading to few melanomas. This suggests that melanin is protective for keratinocytes, but not necessarily for Mcs. Does this mean that Mcs lacking melanin are intrinsically more resistant to transformation than keratinocytes lacking melanin? If so, what are the molecular differences between these 2 cell types? Melanin, and its intermediates, such dopaquinone, and pheomelanin in particular, may damage Mcs, through oxidative stress, for example.
Melanoma susceptibility genes have not been shown to be involved in melanosome transfer. Melanosomes transfer may play no role in melanomagenesis, but this remains an open question, as the molecular mechanisms of transfer have yet to be fully elucidated.
Embryonic development
In mammals, the establishment of the Mcs lineage during embryonic development involves the production of differentiated Mcs, responsible for the pigmentation of skin, hair and fur at birth, and McSC populations, responsible for maintaining pigmentation in the adult.
Normal development. In mice, normal Mcs development starts at approximately embryonic day 8.5 (E8.5), in mid-gestation, when the neural crest cells delaminate from the dorsal part of the closing neural tube and migrate into the MSA. These neural crest cells include the precursors of Mcs, melanoblasts, which proliferate and migrate between the somites (before they become the dermamyotomes, which subsequently evolve into muscle and dermis) and the ectoderm. At approximately stage E10.5, melanoblasts start to express Dct, which serves as a Mcs marker and can be easily detected by X-gal staining in Dct: LacZ mice (106). Between E11.5 and E15.5, the melanoblasts continue to proliferate and migrate through the forming dermis to cover the whole embryo. Some of these melanoblasts cross the basement membrane to colonize the epidermis, before colonizing the future hair follicles. In mice, the melanoblasts give rise to two Mcs populations at birth: the first differentiated Mcs located in the future bulb of the hair, and McSC pool located in the bulge of the hair. During the last steps of Mcs development in the embryo (from E15.5 until E19.5), the melanoblasts begin to express genes encoding enzymes required for melanin production, including Tyr and Tyrp1, which are produced in Mcs and McSC, for a few days after the birth of the McSC (107).
A second wave of melanoblast development has been described in the skin (108). NCCs give rise to several lineages, including Mcs, neurons, chromaffin cells and Schwann cells. One population of engaged precursor cells, the Schwann cell precursors (SCPs), can differentiate into either Schwann cells or Mcs. After early delamination, the SCPs migrate along the ventral pathway, between neural tube and somites, following the nerve fibres. SCPs retain a Schwann cell fate, while they remain in contact with the nerves. In the absence of signals provided by the nerve, some SCPs acquire a melanocytic fate. This second melanoblast population mostly colonizes the dorsal and lateral body walls, and seems to give rise to most of the limb Mcs.
The patterns of congenital pigmentary disorders in humans, including the congenital giant naevi that frequently display NRAS mutations, in particular, helped to identify a third wave of Mcs arising from the ectoderm at the time of gastrulation (109). Temporally, this is actually the first wave, because it occurs before the formation of the neural tube and the NCC formation during embryogenesis. These Mcs are responsible for the non-segmental pattern, through circular, bilateral migration centred on the midline. However, it remains unknown whether these cells contribute to mature Mcs in non-disease states.
Pathological development. Mcs pathology leads to pigmentation disorders of skin and/or hair, and may be associated with deafness and cognitive disorders (110, 111). Waardenburg syndrome (WS) is characterized by pigmentation and hearing disorders, sometimes associated with abnormal development of the face and limbs, and is due to the defective migration and proliferation of embryonic melanoblasts or the abnormal development of other neural crest cells. It is associated with mutations of the MITF and SNAI2 genes (WSII) responsible for the pigmentary and hearing phenotypes; with mutations of the PAX3 gene (WS I and III) affecting neural crest cell development and leading to morphological defects; and with mutations of the EDN3, EDNRB, and SOX10 genes (WS IV) affecting intestinal neural cells. Piebaldism is characterized by hypopigmented patches of skin and hair and is due to the absence of Mcs in certain areas due to defective embryonic/Mcs development. Mutations of the genes coding the KIT receptor and its ligand, SCF, may cause piebaldism syndrome. Apart from MITF, no other melanoma susceptibility genes have been implicated in the embryonic development of Mcs.
Renewal
Normal renewal. McSCs constitute a reservoir for the replacement of Mcs lost during adulthood. McSC niches have been identified in the bulge area of hair follicles (92, 112). McSCs are characterized by a specific cell shape and localization in hair follicles. No specific molecular marker of McSCs has been identified, so a combination of markers is used to follow these cells: McSCs are considered to be Dct-positive, Ki-67-negative, BrdU-retaining cells with low or no expression of KIT and MITF. In the pigment disorder vitiligo, repigmentation often begins at the hair follicles, subsequently spreading out to generate continuous colouring of the skin. This observation is consistent with the notion that McSCs from the bulge can migrate from the hair follicles to the basal layer and differentiate into mature epidermal Mcs. Moreover, repigmentation of depigmented regions lacking hair follicles, such as the palms of the hands, is occasionally observed in patients with vitiligo, indicating that McSC niches are also present in other skin structures, such as sweat or sebaceous glands, and the dermis (113, 114).
The maintenance of hair pigmentation has been well studied in mice and humans. Renewal of the hair in the hair cycle is synchronized with a cycling renewal of the differentiated Mcs, resulting in pigmentation of the new hair. After a resting phase and destruction of the previous hair follicle, the McSCs exit quiescence, proliferate and migrate along the hair follicle as transient amplifying cells, eventually reaching the bulb, where they differentiate into pigment-producing Mcs. Bulge cell quiescence is tightly controlled by several different signals. TGFβ represses differentiation and cell cycle progression; SHH, WNT and β-catenin end the quiescence phase, activating anagen; and NOTCH controls the appropriate differentiation of Mcs (115–121).
Failure of renewal. The absence of Mcs renewal by McSCs can lead to unpigmented skin and hair. The McSC population is limited, despite its potential for renewal, and the number of these cells declines during ageing, resulting in physiological greying of the hair in both humans and mice.
Local depigmentation occurs in adult patients with vitiligo. Vitiligo develops as depigmentation of the skin in specific areas, characterized by a disruption of the epidermal melanin unit with the presence of very few, if any, Mcs. Interestingly, the normally pigmented skin of patients with vitiligo also displays an altered Mcs distribution in the basal layer. The number of keratinocytes between 2 adjacent basal Mcs is larger in the pigmented epidermis of individuals with vitiligo than in that of individuals without vitiligo, and the number of suprabasal Mcs in pigmented epidermis from patients with vitiligo is greater than that in a control population. Alterations to E-cadherin levels at the membrane can affect Mcs adhesion to the basal membrane (105). No melanoma susceptibility gene has yet been linked to renewal of the Mcs lineage or its pathology.
All 20 melanoma susceptibility genes identified to date have a clear function during melanoma initiation, mainly the bypass of senescence. However, except for MITF they do not have a role in the different aspects of the life of Mcs. Susceptibility genes involved in melanogenesis (OCA genes, ASIP, and MC1R), in control of the cell cycle (CDKN2A and CDK4), in telomere length control (TERT, POT1, ACD, and TERF2IP), and in DNA repair (BAP1, RAD51B and POLE) may not be expressed nor have major function during the establishment of the Mcs lineage. As such, we understand that there is no developmental defect associated with the corresponding defective proteins, but we might expect that they may have a role during Mcs maintenance and renewal. Mutations in MITF and KIT dramatically affect the establishment of the Mcs lineage and both proteins play key roles in Mcs development and function. The importance of these genes during renewal remains unclear. Better understanding of the function of all these genes during normal Mcs renewal is crucial for advancing our understanding of their function during melanoma progression, especially during melanoma phenotype switching (86), which may use some proteins involved in McSC biology.
This work was supported by the Ligue Contre le Cancer, INCa, ITMO Cancer, Fondation ARC (PGA), and is under the program “Investissements d’Avenir” launched by the French Government and implemented by ANR Labex CelTisPhyBio (ANR-11-LABX-0038 and ANR-10-IDEX-0001-02 PSL). This work was also supported by a grant from the Icelandic Research Fund (grant number 184861-051 to ES). BBdP is nationwide coordinator of melanoma oncogenetics for INCA.


 Click to show fullsize
Click to show fullsize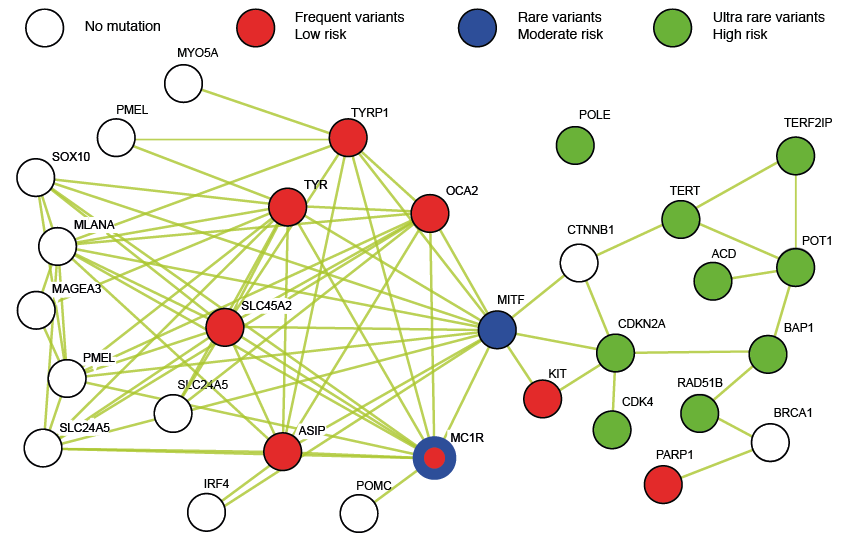 Click to show fullsize
Click to show fullsize