


Department of Dermatology, Le Mans Hospital Center, 194, Avenue Rubillard, FR-72037 Le Mans, France. E-mail: cscard@ch-lemans.fr
Accepted Sep 24, 2020; Epub ahead of print Sep 28, 2020
Acta Derm Venereol 2021; 101: adv00415.
doi: 10.2340/00015555-3645
Muscular toxicity is a well-known and frequent adverse event of statin drugs, affecting an increasing number of patients. The consequence of this toxicity is the discontinuation of statin therapy in 5–20% of cases.
Immune-mediated necrotizing myopathies (IMNMs) are a group of recently described auto-inflammatory myopathies. Three groups have been identified, involving negative autoantibodies, anti-anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMCGR) antibodies, or anti-SRP (anti-signal recognition particle) antibodies. We describe here an association between statin therapy and IMNM involving anti-HMGCR antibodies. The prevalence of this association may vary, as shown by several studies (1, 2).
Prevailing muscular involvement has been described in anti-HMCGR IMNM, with auxiliary damage, such as very rare pulmonary damage (less than 5%) and skin involvement in less than 10% of cases. Skin conditions reported in several studies of anti-HMGCR IMNM include Raynaud phenomenon, a non-specific skin rash, and dermatomyositis-like skin eruptions (3, 4).
We report here a case of a 40-year-old patient with skin disorders that are not classical to IMNM.
A 40-year old man presented with a medical history of myocardial infarction at 37 years of age, with the subsequent introduction of atorvastatin therapy. Aetiological investigations did not identify any coagulation disorder or hereditary lipid abnormalities; he had a history of high blood pressure, dyslipidaemia and active smoking.
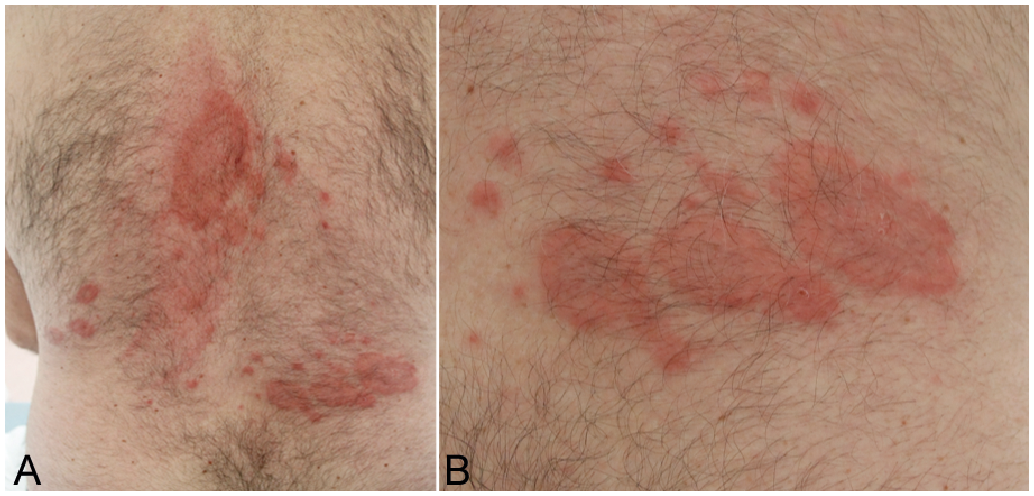
The patient described previous statin intolerance, with muscle pain at each reintroduction of statin therapy and frequent discontinuation, several months before the skin disorder. He showed skin lesions one month before the dermatology consultation, consisting of a non-pruriginous, stable, dermal-type rash, with annular, palpable tender skin lesions and the absence of any other skin lesions. During the consultation, the skin involvement was visible on the upper arms and legs and the back (Fig. 1). It showed progressive extension over several consultations.
Fig. 1. Clinical photographs. (A) Skin presentation on the back. (B) Close-up.
The first diagnostic hypothesis was a benign Jessner lymphocytic infiltrate, confirmed by the pathologist. Benign Jessner lymphocytic infiltrate is a skin disorder, coming as papular erythematous plaques, commonly on the trunk. The main differential diagnoses were lupus erythematosus tumidus, subacute cutaneous lupus erythematosus or Sweet’s syndrome, following the dermal erythematous aspect. Other annular skin diseases could also have been discussed, such as erythema annulare centrifugum or annular sarcoidosis. The histology reported a dermal lymphocytic inflammatory infiltrate, mainly composed of small lymphocytes with histiocytes, with perivascular arrangement. There was no indication of a dermal neutrophilic infiltrate. Direct immunofluorescence was negative. Immunohistochemistry showed an infiltrate consisting of CD3+/CD8+ T lymphocytes without plasmocytes (Fig. 2).
Fig. 2. Histological presentation. (A) Haematoxylin and eosin (x 25) staining. (B) CD3 marking (x 25).
A topical corticosteroid (clobetasol dipropionate 0.05%) and hydroxychloroquine therapy (200 mg twice a day) were introduced without improvement of the skin lesions after one month of treatment. The patient later developed increasing skin involvement and muscle pain. A blood test showed hepatic cytolysis 6 times above the upper limit, predominant alanine transaminases (ALT), and creatine phosphokinase (CPK) 10 times above the upper limit. Aldolase was also increased 10 times above the upper limit. Anti-nuclear antibodies were negative. The patient was hospitalized for cytolysis and muscular pain management. He developed a progressively worsening motor deficit, beginning in the proximal part of the upper and lower limbs, and swallowing disorders, with alimentary wrong ways, dysphagia with solids, dysphonia, and exertional dyspnoea.
Additional investigations found an extended Short T Inversion Recovery (STIR) hyper-signal of the thigh and pelvic muscles on muscular magnetic resonance imaging, consistent with an inflammatory pathology, such as myositis. An electroneuromyogram conducted early did not show a myogenic syndrome: the tracings were normal, except for a depleted tracing in the quadriceps muscle during exercise. Pulmonary function was normal.
A muscular biopsy was performed due to increasing CPK levels, which revealed an unequal diameter of muscle fibres, showing atrophic fibres, with rounded or angular contours, dispersed throughout the muscular spindles, consistent with necrosis and regeneration, and the unusual expression of Class I HLA in the sarcoplasm of the necrotic fibers, suggestive of IMNM. Anti-HMGCR antibodies were positive at > 200 CGU.
Therapy was started, consisting of an intravenous bolus methylprednisolone infusion (250 mg once, then 1 mg/kg/day) because of the swallowing disorders, which rapidly improved, in association with 3 courses of 2 g/kg intravenous immunoglobulin (IVIG) over 2 days. Skin presentation also disappeared within a few days.
Following 3 courses of IVIG associated with physical therapy, the patient showed a decrease in CPK and aminotransferase levels, associated with an increase in muscular mass.
Faced with a slow decrease in symptom severity, despite high-dose corticosteroid treatment, and the recurrence of smaller skin lesions, mycophenolate mofetil therapy (1,000 mg twice a day) was introduced, leading to a rapid decrease in muscle and skin symptoms and a return to normal hepatic enzyme and CPK levels by 28 days after the beginning of treatment.
IMNMs are rare diseases, with a prevalence of only 9–14 cases per 100,000 people (5, 6) and only ~10% involve either anti-SRP or anti-HMGCR myopathy (7). Anti-HMGCR autoantibodies were discovered in 2010 by screening serum from “autoantibody-negative” necrotizing myositis patients (3).
Skin involvement has been described in less than 10% of cases and is generally dermatomyositis-like. There are no findings of Jessner-like T-cell infiltrates in anti-HMGCR IMNM reported in the literature. There have also been no cases showing large erythematous lesions in dermatomyositis-like skin disorders similar to that described in this case report (8).
Pathology findings in dermatomyositis cases show slight hyperkeratosis and epidermal atrophy, with effacement of the rete ridge and vacuolar degeneration of basal keratinocytes. There is also upper dermal oedema and, sometimes, the presence of melanophages. Perivascular lymphocytic infiltration of activated T lymphocytes and macrophages is present. Dermal mucin deposits are suggestive of dermatomyositis. Direct immunofluorescence of lesional skin may reveal granular deposits of immunoglobulin and complement at the dermal–epidermal junction. The dermal infiltrate in the skin of our patient can be likened to that of dermatomyositis, without clinical comparison.
This case is unusual because of the uncommon clinical dermatological presentation of this type of IMNM, like that of Jessner-Kanoff disease. There was no delay in provision of appropriate care for this patient because of the early muscular symptoms, which were evocative of a myopathy.
This unusual clinical presentation can serve as the basis for discussion of skin disorders associated with immune-mediated necrotizing myopathies, which are rare, show a variable functional prognosis, and require extended immunosuppressive therapy.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize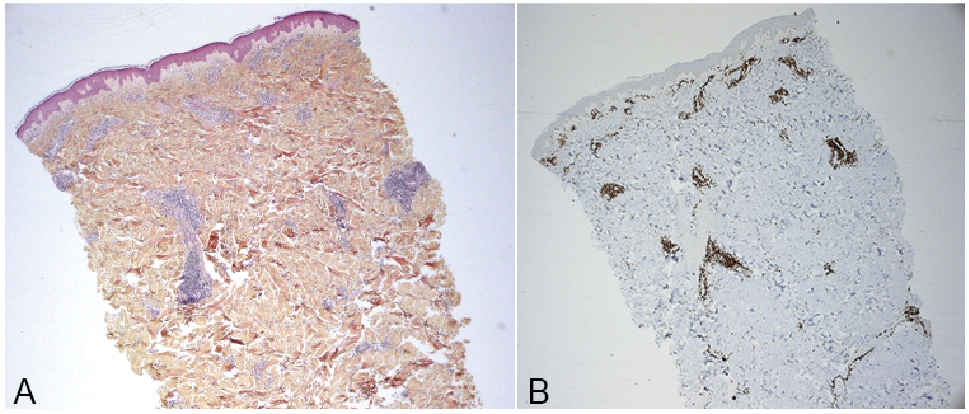 Click to show fullsize
Click to show fullsize