Atsushi Tokuriki, Takahiro Kiyohara, Toshiko Ido and Masanobu Kumakiri
Department of Dermatology, Faculty of Medical Sciences, University of Fukui, 23-3 Matsuoka-Shimoaizuki, Eiheiji, Fukui 910-1193, Japan. E-mail: kiyo@u-fukui.ac.jp
Accepted November 7, 2011.
Primary cutaneous CD30+ T-cell lymphoproliferative disorders include: (i) lymphomatoid papulosis (LyP), (ii) primary cutaneous anaplastic large cell lymphoma (pcALCL), and (iii) borderline lesions (1). LyP represents the benign end of this spectrum of disorders; however, some cases are associated with progressive cutaneous lesions and/or extracutaneous involvement (2). Although pcALCL usually has a favourable prognosis, extensive limb disease (ELD) is believed to indicate a poorer prognosis (3). We report here the case of a man who presented with ELD, extracutaneous involvement, and acquired ichthyosis during the typical course of LyP.
CASE REPORT
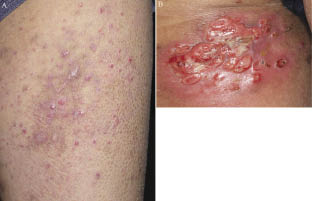
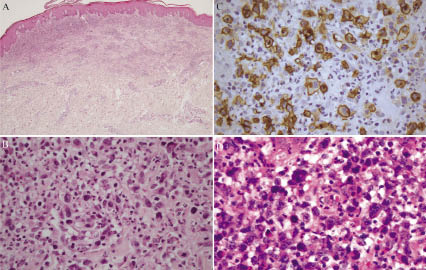
A 41-year-old man presented with self-healing, but recurrent, erythematous papules on the face, trunk, and extremities, which were first noted when he was 28 years old (Fig. 1A). Histological examination revealed a typical picture of LyP, with a dermal wedge-shaped cell infiltrate (Fig. 2A), which comprised anaplastic large cells in an inflammatory background. The tumour cells had an anaplastic morphology with irregular-shaped nuclei, prominent nucleoli, and abundant cytoplasm (Fig. 2B). Immunohistochemically, they were diffusely positive for CD30 (Fig. 2C), partially positive for CD3 and CD4, but negative for CD8, CD15, CD20, CD25, anaplastic lymphoma kinase, and epithelial membrane antigen. Also, he had exhibited ichthyosiform eruptions on the extremities and abdomen since he was 40 years old (Fig. 1A). His family members had no history of similar skin symptom. The histology showed orthokeratotic hyperkeratosis with a decreased granular cell layer. All together, we diagnosed the patient as LyP associated with acquired ichthyosis. He had been treated with a topical steroid agent and phototherapy (PUVA), but did not achieve complete remission of the papules. The ichthyosiform eruptions improved slightly with 20% urea ointment. At the age of 42 years, left inguinal lymphadenopathy abruptly developed. Histologically, the aggregation of anaplastic large cells with the same phenotype as those of papules in the cortex was observed. Radiation therapy (total radiation dose: 46 Gy/cm2) was performed, resulting in temporal remission. At the age of 45 years, an ulcerative, erythematous tumour abruptly developed on proximal and flexor sites of the left thigh, followed by recurrent left inguinal lymphadenopathy. The tumour was excised, but multiple tumours soon developed on the operation scar (Fig. 1B). Histologically, the tumour mass was composed mainly of anaplastic large tumour cells (Fig. 2D) involving the whole dermis. The tumour cells showed a phenotype identical to those of the papules and lymph nodes. Southern-blotting analysis of T-cell receptor (TCR) Cβ1 and Jγ genes in the tumour showed no clonal TCR gene rearrangement, as with the papules and lymph node. Imaging tests detected no further systemic involvement. We re-started local radiation therapy, and the lymph node was sensitive to irradiation (total radiation dose: 28 Gy/cm2). The tumours were also sensitive to radiation; however, new tumours continuously developed in surrounding, non-irradiated areas over approximately a 3-month period. Finally, the tumours expanded to the left popliteal fossa, and thus we decided to increase the irradiated areas (total radiation dose: proximal site: 64 Gy/cm2, distal site: 32 Gy/cm2). We commenced treatment with low-dose (15 mg/week) methotrexate (MTX). The tumours responded well to MTX and stopped developing approximately 2 weeks after MTX introduction. Recently, we have tapered MTX (15 mg/2 weeks), with a few papules remaining; however, no new skin tumours and systemic lesions have been found.
Fig. 1. (A) Scattered red papules and ichthyosiform eruptions on the left thigh. (B) Grouped ulcerated tumours, 5 to 23 mm in diameter, on the proximal and flexor sites of the left thigh.
Fig. 2. (A) A papule shows wedge-shaped cell infiltrate in the dermis (haematoxylin-eosin staining × 100). (B) The cell infiltrate is composed of anaplastic large cells in an inflammatory background. (C) Immunohistochemical staining of a papule. Large cells are positive for CD30. (D) Anaplastic large cells proliferate in the tumour. (haematoxylin-eosin staining × 400).
DISCUSSION
LyP was referred to as a benign, skin-limited, chronic disease in its original description (4). However, Bekkenk et al. (2) have indicated that approximately 20% of patients with LyP are associated with lymphomas, and 10% of those are associated with extracutaneous involvement. Most patients with pcALCL present with solitary or localized tumours, classified as T1 or T2 according to the ISCL-EORTC TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sézary syndrome (5). The prognosis is usually favourable, with a 10-year disease-specific survival of approximately 90% (6). However, Liu et al. (7) indicated that a subset of patients with pcALCL with ELD might follow a more aggressive clinical course. Patients with ELD were noted to present with extensive tumour involvement of a single limb, classified as T2b or T2c, or a single limb with contiguous body regions, classified as T3b. Woo et al. (3) have reported a study of 48 patients with pcALCL, including 4 with ELD. This study also included 8 patients with pcALCL with extracutaneous involvement. Univariate analysis demonstrated that ELD and extracutaneous involvement were significant prognostic factors for the 5-year disease-specific survival. The clinical course indicated that our case was a poor one in which LyP progressed to pcALCL, classified as T2c. Although no monoclonal rearrangement of TCR genes has been identified, they should be regarded as ELD because of the aggressive clinical course. In the study by Woo et al. (3), 4 patients with ELD were characterized by the lack of a durable response to treatment. All but one with ELD progressed regionally within 3 months of completing chemotherapy or local radiation. They also failed to salvage treatments, including various chemotherapy regimens. Our case was similar to their cases, in that ELD progressed regionally over an approximately 3-month period in spite of local radiation, but was different, in that low-dose MTX therapy was effective and no new skin tumours and systemic lesions have been found for approximately 3.5 years.
Acquired ichthyosis is a rare and significant manifestation, as it is generally associated with underlying lymphoproliferative disorders, such as Hodgkin’s disease (8). Yokote et al. (9) demonstrated a case of LyP preceded by ichthyosis. Their case was similar to ours, in that LyP was followed by inguinal lymphadenopathy. Morizane et al. (10) reported that, of 10 patients with pcALCL, 3 (30%) were associated with ichthyosis. All 3 cases of PC-ALCL with ichthyosis were in the advanced stages and showed internal lymph node involvement. In our case, inguinal lymphadenopathy initially occurred 2 years after the onset of ichthyosis, followed by ELD and the recurrence of lymphadenopathy. Overall, we hypothesize that LyP or pcALCL patients with acquired ichthyosis have a poorer prognosis than those without ichthyosis. Thus, more careful follow-up may be required when identifying ichthyosis during the course of LyP or pcALCL.
REFERENCES