


Department of Dermatology, University Erlangen, Ulmenweg 18, DE-91052 Erlangen, Germany. *E-mail: Michael.Erdmann@uk-erlangen.de
Accepted Apr 18, 2017; Epub ahead of print Apr 19, 2017
Scleromyxoedema was first described by Dubreuilh (1) & Reitmann (2) as lichen myxoedematosus. Also known as Arndt-Gottron disease, this disorder belongs to the group of primary cutaneous mucinoses and presents as a generalized waxy, papular eruption, usually accompanied by monoclonal gammopathy. Without therapy, the dermal mucinosis spreads and systemic manifestations can occur, resulting in significant morbidity and mortality. We report here a case of scleromyxoedema in a young woman, which initially mimicked a photodermatosis that responded to intravenous immunoglobulin (IVIG) therapy.
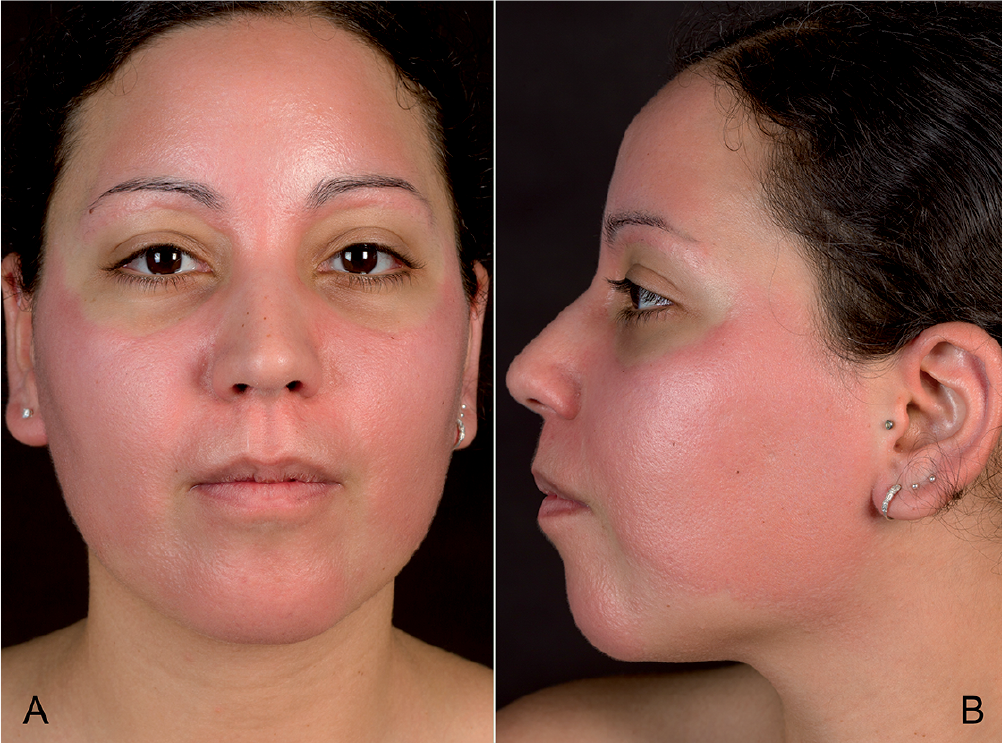
A 32-year-old woman initially presented to her local dermatologist with facial erythema after a holiday in Egypt. Within several days the erythema had spread to her upper extremities and neckline. Consistent with photodermatosis, the erythema was strictly limited to areas of sun exposure, omitting the periorbital region, the skin beneath the chin, and 2 linear areas over the temples (Fig. 1). A biopsy showed chronic eczema and the physician diagnosed a dermatitis solaris, which was treated with topical glucocorticoids without improvement. Because of spreading and continuous induration of the eruption, the patient presented at our department.
Fig. 1. (a) Frontal and (b) profile view of the face showing erythema omitting the periorbital region and over the temples. A written permission is given by the patient to publish these photos.
She described limited mobility and rigid facial expression with difficulties in opening her mouth. Light provocation and patch testing for contact eczema did not reveal any pathological findings. Blood testing showed monoclonal gammopathy of immunoglobulin G with predominant lambda light chains. Tests for antinuclear antibodies and myositis-associated autoantibodies were negative. Capillaroscopy revealed a normal capillaroscopic pattern. Haematoxylin and eosin (H&E) and Alcian blue staining of a skin biopsy revealed prominent and diffuse mucinosis in the upper dermis with increased collagen deposition and irregularly arranged fibroblasts (Fig. 2) with no histological evidence of cutaneous lupus erythematodes or dermatomyositis. Based on the clinical and histological findings and histology, scleromyxoedema was diagnosed.
Fig. 2. Alcian blue staining revealing prominent and diffuse mucinosis in the upper dermis with increased collagen deposition and irregularly arranged fibroblasts.
We initiated therapy with 200 mg hydroxychloroquine administered 3 times daily, which led to waning of the erythema. However, the induration and coarseness of the skin increased. Therefore, we began therapy with IVIG (2 g/kg body weight, every 4 weeks). This treatment rapidly improved the appearance of the skin, and an almost complete resolution was observed after 3 cycles of IVIG (Fig. 3). Remission is still ongoing under continuation of IVIG (1.5 g/kg body weight, every 4 weeks). Further reductions in the dose led to recurrent disease.
Fig. 3. Almost complete resolution of the scleromyxoedema after 3 cycles of intravenous immunoglobulins. A written permission is given by the patient to publish this photo.
Diagnosis of scleromyxoedema is based on a generalized papular and sclerodermoid eruption, monoclonal gammopathy, absence of a thyroid disorder, and the histological triad of mucinosis, fibrosis, and fibroblast proliferation (3). Scleromyxoedema can mimic systemic scleroderma, with symptoms such as sclerodactyly, oesophageal dysmotility, and Raynaud’s phenomenon. Characteristic distribution of papules at the glabella, posterior auricular area, and middle portion of the back of scleromyxoedema are absent in scleroderma (4). The typical posterior auricular and glabellar involvement was not observed in our patient, which may be explained by the early diagnosis and treatment. In addition, the IgG monoclonal gammopathy favours diagnosis of scleromyxoedema when distinguishing between scleroderma and nephrogenic systemic fibrosis (NSF). In contrast to scleromyxoedema, fibroblast proliferation is absent in scleroderma; nevertheless, mucin deposition can be seen between collagen bundles.
The pathogenesis of scleromyxoedema and the significance of the accompanying monoclonal gammopathy are not fully understood. The stimulation of glycosaminoglycan synthesis and fibroblast proliferation by circulating cytokines, such as interleukin-1, tumour necrosis factor (TNF)-α, and transforming growth factor-beta, has been suggested (5, 6). The origin of these cytokines is attributed to the bone marrow because autologous stem cell transplantation leads to clinical remission in some cases (7).
At 32 years of age, our patient was 2 years younger than the youngest patient in a multicentre study of 30 patients, with a mean age of 59 years and no sex predominance (8). At first presentation, no extracutaneous manifestations were observed. In 30% of the patients, neurological features, such as carpal tunnel syndrome or involvement of the central nervous system (e.g. vertigo, stroke, seizure, and psychosis) can be seen (8). In addition, rheumatological symptoms, such as arthralgia and cardiovascular abnormalities with heart failure, myocardial ischaemia, and heart block, occur in up to 25% of the patients (8). More uncommon gastrointestinal (e.g. oesophageal dysmotility and dysphagia), respiratory (e.g. dyspnoea and decreased epiglottis and vocal cord mobility), renal, and ocular symptoms can also be present.
Due to lack of randomized trials, the efficacy of several known therapies has been evaluated only in case series. Therapy with IVIG is considered as the treatment of choice, with a favourable toxicity profile and high efficacy, especially in refractory cases and patients with systemic manifestations. It is suggested that the immunomodulatory effects of IVIG lead to neutralization of circulating cytokines that play a role in scleromyxoedema (3, 8). However, IVIG treatment is expensive and time consuming. It is administered over a period of several days at a dose of 2 g/kg body weight every 4 weeks. Nevertheless, there is still debate about the appropriate dose, since patients also benefit from lower doses (1 g/kg) (3). Patients who do not respond to IVIG may benefit from thalidomide, TNF-α blockers, glucocorticoids, retinoids, interferon-alpha, cyclophosphamide, cyclosporine, plasmapheresis, autologous stem cell transplantation, or radiotherapy. However, these therapeutic options often have severe adverse effects, particularly in long-time usage (3, 8, 9). The relevance of monoclonal gammopathy is much debated, especially whether specific treatment is required considering the side-effects. However, upon failure and/or intolerance of above mentioned therapies, chemotherapy of gammopathy may be initiated.
Scleromyxoedema is an unpredictable and usually progressive disease, which can be disabling and cause high mortality in the absence of successful treatment. The rate of recurrence is very high after withdrawal of therapy; therefore, long-term maintenance treatment and follow-up are necessary.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize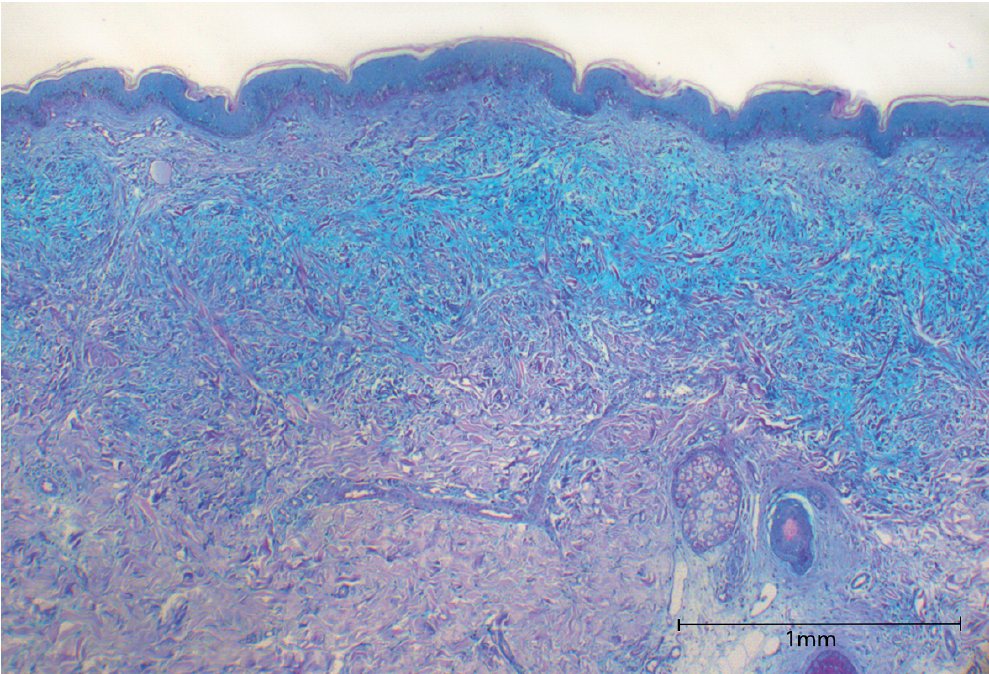 Click to show fullsize
Click to show fullsize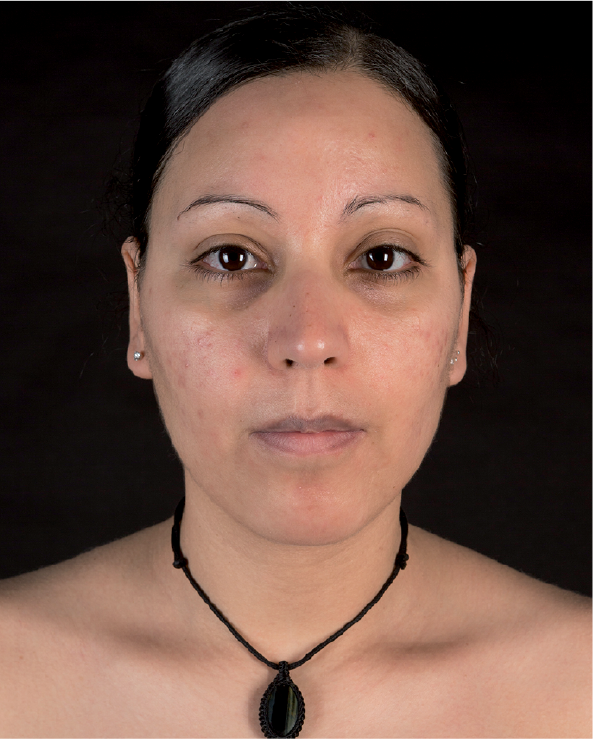 Click to show fullsize
Click to show fullsize