


1Department of Dermatology, 5Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 80708, 2Graduate Institute of Clinical Medicine, College of Medicine, 4Department of Anatomy, College of Medicine, Kaohsiung Medical University, Kaohsiung, 3National Institute of Environmental Health Sciences, National Health Research Institutes, Miaoli County, Taiwan. *E-mail: epor324@gmail.com and ylhsieh@kmu.edu.tw
#These authors contributed equally.
Accepted Apr 27, 2017; Epub ahead of print Apr 27, 2017
Sjögren’s syndrome (SS) is an autoimmune disorder characterized by exocrine dysfunction, such as xero-phthalmia or xerostomia, focal lymphocytic sialoadenitis, and a high titre of autoantibodies inclusive of anti-Ro and anti-La. SS is also associated with both organic dermatological disorders and neuropathy. Dry skin is a complication in 23–67% of patients with SS, especially those under 50 years of age (1). Two cases of acquired generalized hypohidrosis/anhidrosis (AGHA) associated with subclinical SS and absence of dryness of the eyes and mouth have been described previously (2, 3). In addition, peripheral neuropathy, the spectrum of which includes autonomic neuropathy, is the most common neurological complication of SS, with a prevalence ranging from less than 2% to greater than 60% (4). Autonomic neuropathy may cause symptoms such as orthostatic hypotension, bowel dysfunction and anhidrosis. We report here a case of subclinical SS-associated AGHA.
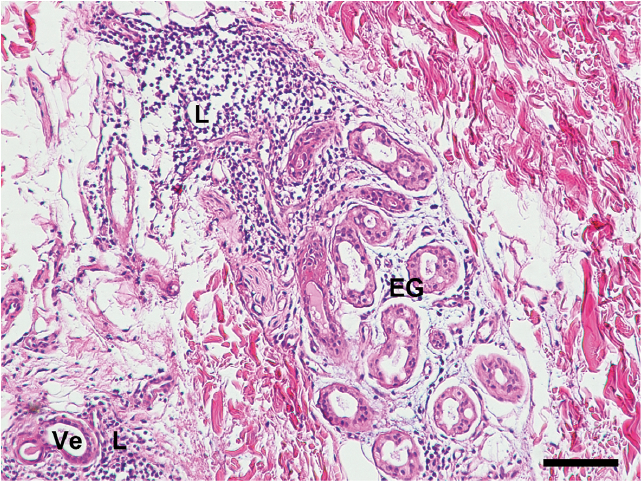
A 35-year-old man experienced an episode of dry, hot sensation and fever up to 39°C after cycling. No chills, muscle soreness, dizziness, skin rash, itch or infections were noted at that time. Thereafter, persistently generalized decreased sweating was noted for 1 month. The patient had previously been healthy and his family history was unremarkable. He visited our outpatient department and hypohidrosis/anhidrosis was confirmed with a starch-iodine test, showing decreased or absent sweating on the face, trunk and extremities. In addition, intradermal injection of 1% pilocarpine did not elicit sweating. Laboratory findings revealed elevated anti-Ro (> 240 EliAU/ml, normal < 7 EliAU/ml), anti-La (190 EliAU/ml, normal < 7 EliAU/ml), antinuclear antibody (1:40), anti-extractable nuclear antigen (ENA > 32, normal < 0.7), anti-microsomal antibody (331 IU/ml, normal < 35 IU/ml), carcinoembryonic antigen (CEA, 310 ng/ml, normal: 0~5 ng/ml), serum IgG (1,960 mg/dl, normal: 700~1,600 mg/dl) and IgE (153.0 IU/ml, normal < 87 IU/ml). Serum complement C3 was reduced (72.5 mg/dl, normal: 90~180 mg/dl). Anti-RNP, anti-Sm, anti-dsDNA, anti-Scl 70 antibodies, antiphospholipid antibodies (APL) IgG/IgM, anticardiolipin (aCL) IgG/IgM, rheumatoid factor, creatine phosphokinase (CPK), anti-mitochondrial antibody, HBs antigen, anti-HCV IgG and HIV test were negative. A complete blood cell count, renal function, alkaline phosphatase (ALP), bilirubin level, urinalysis, stool routine, were within normal limits, except for a slight elevation of aspartate aminotransferase (AST) and alanine transaminase (ALT), which might be due to certain inflammatory process. Salivary scintigraphy uncovered mild sialoadenitis and reduced salivary excretory function. Schirmer’s test was unremark-able. Brain magnetic resonance imaging did not reveal any notable abnormality. Electromyography revealed abnormal sudomotor sympathetic function. A nerve conduction velocity test disclosed mild bilateral median motor neuropathy and mild right ulnar sensory neuropathy. Incisional biopsy of anhidrotic skin showed lymphocytic infiltration in the vessels of the upper dermis and the eccrine glands (Fig. 1). No obvious structural abnormality was found in the epidermis, dermis and appendages, including sweat glands. Direct immunofluorescence was unremarkable. electron microscopy (EM) study of the sweat gland showed macrophage phagocytosed unmyelinated nerve bundle (Remak bundle, Fig. 2A) and the profiles of collagen pockets on Remak bundle (Fig. 2B), indicating the degeneration of sympathetic efferents to eccrine glands. The patient declined a labial biopsy.
Fig. 1. Histopathology of anhidrotic skin. Biopsy showing eccrine glands (EG) and vessels (Ve) infiltrated by lymphocytes (L), indicting that skin inflammation underlies the anhidrosis observed in a patient with subclinical Sjögren’s syndrome. (Haematoxylin and eosin staining. Bar: 100 µm).
Fig. 2. Ultrastructural analysis of the patient’s sweat gland indicates degeneration of unmyelinated sympathetic fibre innervating the gland. (A) Macrophage phagocytosed bundle of unmyelinated nerves (Remak bundle). (B) The remains of degenerated Remak bundle and axonal space were occupied by collagen fibres, termed collagen pockets. (M: macrophage; R: Remak bundle; CP: collagen pocket; SN: nucleus of Schwann cells. Bar: 2.5 μm).
It was considered that the patient’s anhidrosis might be associated with subclinical SS, in that no apparent dryness of the mouth or eyes could fully conform to diagnostic criteria. Sweating increased after intravenous steroid pulse therapy (methylprednisolone 250 mg every 12 h for 3 days), and remained stable for the following 8 months. The CEA level decreased significantly after treatment, from 310 ng/ml to 72 ng/ml at 3 weeks, and to 46 ng/ml at 8 months.
It has been suggested that a possible mechanism behind generalized anhidrosis associated with SS is an acquired anomaly of the sweat glands (3). In our patient, no remarkable structural abnormality was noticed, except for prominent lymphocytic infiltration around the vessels and eccrine glands. Eccrine gland dysfunction, with both impaired direct sweating and axon reflex-mediated sweating, has been reported in a patient with SS (5), which might be due to autoimmune mechanisms mediated by T cells or muscarinic acetylcholine receptor M3 autoantibodies (3, 6). Absent reaction in the 1% pilocarpine intradermal test demonstrated dysfunction of the sweat glands. In our patient, EM identified denervation of sympathetic-innervating eccrine glands, consistent with the clinical findings of abnormal sudomotor sympathetic function. Decreased innervation of the eccrine glands accompanied by an impaired nerve conduction velocity test substantiated neural involvement of the patient’s AGHA. These findings therefore suggested that both eccrine dysfunction and neural abnormality are involved in the mechanisms of subclinical SS-associated AGHA, which is contrary to the previous reports of Katayama et al. (1). Intriguingly, Goto et al. reported a case of primary SS with AGHA whose skin biopsy showed atrophic changes in the glands without infiltrating inflammatory cells and EM findings including atrophic glands and reduction in the number of nerve terminals and unmyelinated axons around the secretory coils (7). The current case further strengthens the theory that SS or subclinical SS-associated AGHA is related to inflammation in both sudomotor fibres and sweat glands, as demonstrated by functional nerve conduction studies, skin histopathology and EM.
Previous reports have reported a concurrent urticarial episode in some patients with acquired idiopathic generalized anhidrosis (AIGA) (8), but not in patients with SS-associated AGHA, as demonstrated in the current case. Neural involvement might be a possible explanation. Sudomotor sympathetic nerve activity is well preserved or even increased in AIGA (9), and therefore mast cells close to eccrine glands are activated by overflowing acetylcholine releasing from nerves, leading to histamine release (8). However, such mechanism may be obstructed owing to peripheral neural damage in SS-associated AGHA. It is suggested that mast cell activation by other neuropeptides, such as substance P, vasoactive intestinal peptide, neuropeptide Y and calcitonin gene-related peptide (10), are reduced in SS-associated AGHA, although further research is needed.
Serum CEA has been suggested as a clinical marker to reflect the disease activity of AIGA (11). Our patient demonstrated a concurrent improvement in sweating and serum CEA after therapy, suggesting CEA as a potential clinical indicator of subclinical SS-associated AGHA. There is currently no consensus about treatment of SS-associated AGHA (4, 5). Steroid pulse therapy or high-dose oral steroids are widely used in the treatment of AIGA (12). In our patient, remission without recurrence at 8 months was achieved after a single course of methylprednisolone, 250 mg every 12 h for 3 days, without continuing oral steroids. The presence of perieccrine inflammation in sweat gland dysfunction seemed to respond well to systemic treatment with corticosteroid. Maeda et al. described a case of SS-associated anhidrosis treated successfully with oral prednisolone (5). Moreover, cyclosporine and intravenous immunoglobulin are also potential therapeutic alternatives for treatment of SS-associated AGHA (3, 13).


 Click to show fullsize
Click to show fullsize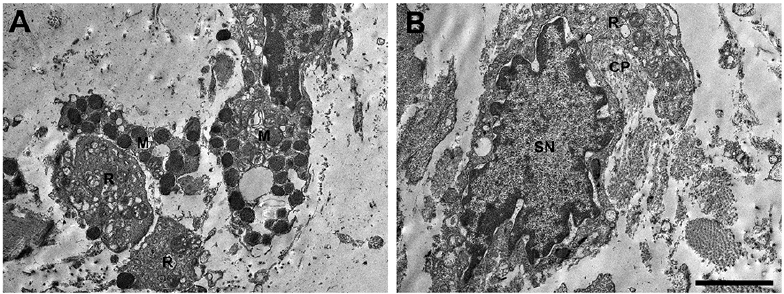 Click to show fullsize
Click to show fullsize