


1Department of Dermatology, Iwate Medical University School of Medicine, 19-1 Uchimaru, Morioka, Iwate 020-8505, 2Division of Dermatology, Kitakami Saiseikai Hospital, Iwate, and 3Department of Pediatrics, Tohoku University School of Medicine, Miyagi, Japan. E-mail: dwatabe@iwate-med.ac.jp
Accepted Apr 26, 2018; Epub ahead of print Apr 27, 2018
Holocarboxylase synthetase deficiency (HCSD) is a rare autosomal recessive disorder of biotin metabolism (1). Holocarboxylase synthetase (HCS) plays an essential role in biotin utilization in cells, and its deficiency causes biotin-responsive multiple carboxylase deficiency (MCD) in humans (2). Most patients with HCSD develop symptoms within the first few days or first 2 months of life. The clinical symptoms include tachypnoea, feeding difficulties, seizures, and dermatitis in the early infant period. We report here a case of HCSD presenting as persistent psoriasis-like dermatitis in adulthood.
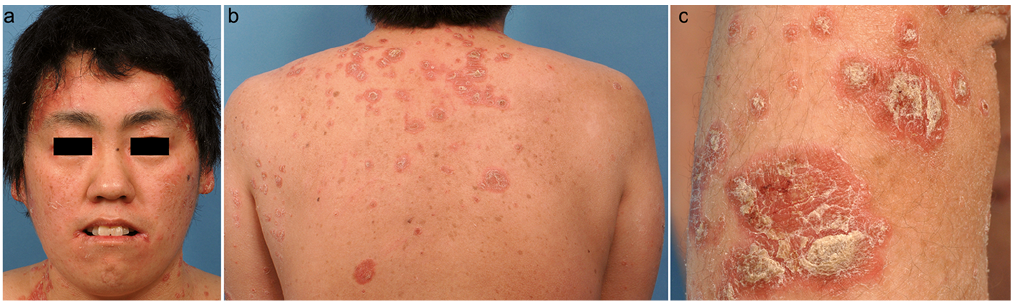
A 34-year-old woman presented with a skin eruption covering the whole body that had persisted since infancy. She had been born to healthy, unrelated Japanese parents. The patient had developed tachypnoea and myoclonic seizures from the second day of life (3). Investigation confirmed severe metabolic acidosis and ketosis. The results of urinary organic acid analysis and fibroblast carboxylase tests were consistent with a diagnosis of HCSD. With biotin therapy (20 mg/day), her condition had improved quickly, although intermittent respiratory infections had led to metabolic acidosis and organic aciduria. Gene mutation analysis revealed p.L237P and c.780delG (formerly termed delG1067) in the heterozygous form (2). Skin lesions had developed from 6 months after birth, and she had been treated with oral retinoid for a diagnosis of psoriasis from 10 years of age. She had no family history of psoriasis. Her recent medication included biotin (DSM Nutrition, Tokyo, Japan) and L-carnitine supplementation, at 50–100 mg/day and 1,000 mg/day, respectively. Physical examination at our hospital revealed a well-demarcated scaly erythematous plaque on the scalp, face, trunk and extremities (Fig. 1). Involvement of the intertriginous area and angle of the mouth was also evident. The nails were unremarkable, and the hair had a normal texture with no areas of alopecia. Histological examination of a skin biopsy specimen revealed hyperkeratosis, elongation of the rete ridges, and neutrophilic infiltration below the stratum corneum, being compatible with psoriasis (Fig. S1a, b). Based on these findings, the skin lesions were considered to be consistent with psoriasis-like dermatitis of HCSD. Oral retinoid and topical betamethasone and calcipotriol had not ameliorated the skin lesions sufficiently, and there had been repeat exacerbation with respiratory tract infection.
Fig. 1. Well-demarcated scaly erythematous plaque on (a) the face, (b) the back and (c) the forearm. Permission from the patient is given to publish these photos.
The patient was admitted 4 years later with upper respiratory tract infection and high fever. Clinical examination showed an erythematous plaque with pustules and scales on the face, trunk and extremities (Fig. 2). Histological examination of a skin biopsy specimen revealed an extensive neutrophilic infiltration with marked spongiform pustules (“Kogoj’s microabscesses”) below the stratum corneum (Fig. S2). Laboratory examinations yielded elevation of the serum C-reactive protein level 5.92 mg/dl (normal < 0.3 mg/dl), and the serum zinc level was normal. The patient was treated with intravenous ampicillin, and the fever and skin lesions resolved gradually.
Fig. 2. Erythematous plaques with pustules and scales on (a) the face, (b) the abdomen and (c) the back. Permission from the patient is given to publish these photos.
HCSD is an autosomal recessive disorder of organic acid metabolism in humans (1). HCS catalyses the transfer of biotin to 4 biotin-dependent enzymes: 3-methylcrotonyl CoA carboxylase, propionyl CoA carboxylase, pyruvate carboxylase, and acetyl CoA carboxylase. Deficient HCS activity results in reduced activity of multiple carboxylases (2). The diagnosis is confirmed by mutation analysis or by measurement of carboxylase activities in fibroblasts at low or high concentrations of biotin. The skin manifestations of HCSD include a sharply marginated, seborrhoeic rash on the scalp, eye-brows, and eyelashes, which can spread to the perioral, perinasal, and other flexural areas (4). Hair thinning and alopecia are also evident. Few previous reports have detailed the skin manifestations of HCSD. Most case reports have focused on metabolic or molecular analysis of HCSD. We have reviewed previous case reports of HCSD describing skin lesions (Table SI). Among them, the most common symptoms described were alopecia, periorificial and intertriginous dermatitis. Seborrhoeic dermatitis, psoriatic dermatitis and ichthyosis are also seen in some patients. All of these skin manifestations have been observed in neonates and children, but this was not the case in our patient.
To our knowledge, this is the first report of a patient with HCSD presenting with a persistent skin lesion in adulthood. Most patients with HCSD have a good prognosis if biotin therapy is introduced early. Thus, only severe cases would show clinical symptoms, such as skin lesions, in adulthood. In our present patient, the maximal activity of HCS was very low, and she sometimes developed ketoacidosis due to respiratory infection despite administration of high-dose biotin (50–100 mg/day). In addition, along with the exacerbation of HCSD, the patient developed skin symptoms. Therefore, we considered the psoriasis-like dermatitis to be a skin manifestation of HCSD, rather than a complication of psoriasis.
The pathogenesis of skin manifestations in HCSD has not been fully explained. It has been postulated that a defect of fatty acid synthesis due to reduced activity of acetyl CoA carboxylase is implicated in the skin manifestations of MCD and biotin deficiency (5). Nakajima et al. (6) reported that newborn serine palmitoyltransferase (SPT)-knockout mice showed significantly decreased epidermal levels of ceramide. They then developed alopecia and psoriasis-like skin lesions mediated by interleukin (IL)-23-dependent γδ T cells, which produce IL-17 and IL-22. Therefore, ceramide deficiency resulting from a defect of fatty acid synthesis may lead to psoriasis-like lesions in HCSD, although the exact mechanism remains to be determined.
Recent studies have shown that biotin also regulates immunological and inflammatory function. Agrawal et al. (7) reported that biotin deficiency facilitates the secretion of tumour necrosis factor (TNF)-α, IL-1, IL-23, and IL-12p40 from dendritic cells, and that biotin-deficient dendritic cells induce significantly higher levels of secretion of IFN-γ, IL-17, and IL-22 from CD4 T cells. These findings suggest that biotin deficiency leads to a shift of Th-cell responses towards Th1/Th17. Among the various skin disorders, the IL-23/Th17 pathway (8) and the IL-1/IL-36 axis (9) have been considered to play a major role in the pathogenesis of psoriasis and pustular psoriasis, respectively. The skin manifestations of HCSD may also be attributable to these immunological alterations.
Psoriasis is a chronic inflammatory systemic disease. Care should be taken to ensure that chronic inflammation of persistent skin lesions does not lead to comorbidities, such as arthritis, inflammatory bowel disease (IBD), uveitis, metabolic syndrome, and psychological or psychiatric disorders.
Further studies and case reports are required to clarify the pathogenetic changes occurring in the skin of patients with HCSD.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize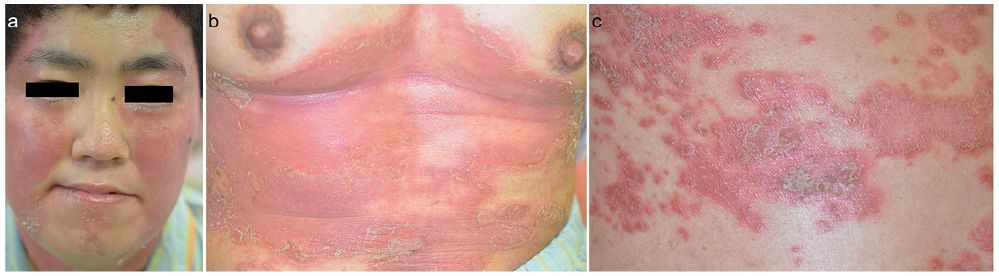 Click to show fullsize
Click to show fullsize