


1Department of Medicine, Dermatology and Venereology Unit, Karolinska Institutet, and 2Unit of Dermatology, Karolinska University Hospital, Stockholm, Sweden
Tofacitinib is a Janus kinase (JAK) inhibitor, which has shown efficacy in treating psoriasis. The mode of action of tofacitinib is not completely understood but it has been thought to be mediated by the inhibition of CD4+ T-cell activation. Here, we investigated whether the molecular targets of tofacitinib are expressed in keratinocytes, and whether tofacitinib can modulate the activity of the JAK/Signal Transducer and Activators of Transcription (STAT)-pathway in keratinocytes. Transcriptomic profiling of human keratinocytes treated with IL-22 in combination with tofacitinib revealed that tofacitinib could prevent the majority of IL-22-mediated gene expression changes. Pathway analysis of tofacitinib-regulated genes in keratinocytes revealed enrichment of genes involved in the JAK/STAT signalling pathway. Quantitative real-time-PCR confirmed the upregulation of S100A7 and downregulation of EGR1 expression by IL-22, which was prevented by tofacitinib pre-treatment. These results indicate a direct effect of tofacinitib on keratinocytes, which can have relevance for systemic as well as for topical treatment of psoriasis with tofacitinib.
Key words: JAK-STAT (Janus kinase-signal transducer and activators of transcription); tofacitinib; keratinocyte; psoriasis; cytokines.
Accepted May 4, 2018; Epub ahead of print May 8, 2018
Acta Derm Venereol
Corr: Enikö Sonkoly, Dermatology and Venereology Unit, Department of Medicine Solna, Karolinska Institutet, CMM L8:02, SE-17176 Stockholm, Sweden. E-mail: eniko.sonkoly@ki.se.
Psoriasis is the most common chronic inflammatory skin disease in adults. Tofacitinib is an immunomodulating drug that has shown effect for the treatment of psoriasis in clinical trials. The mode of action through which tofacitinib exerts its effects is not fully understood. Previous studies reported that the effects of tofacitinib are mediated by the inhibition of T-cell activation. In this study we found that tofacitinib can efficiently repress inflammatory pathways in skin cells (keratinocytes), indicating that the effect of topical or oral tofacitinib treatment can partially be mediated via keratinocytes in addition to immune cells.
Psoriasis is a chronic immune-mediated skin disease that affects 2–3% of the population worldwide (1). The Janus kinase/signal transducer and activators of transcription (JAK/STAT) pathway is central in the pathogenesis of psoriasis (2), as evidenced by genetic association of STAT3 with psoriasis (3), and the essential role of STAT3 in psoriasis-related cytokine signalling pathways (interleukins [IL]-6, IL-10, IL-20, IL-22, and IL-23), and in differentiation of Th17 cells, a key cell type in psoriasis (3, 4). Tofacitinib (CP-690,550), a potent inhibitor of JAK1 and JAK3, and to some extent to JAK2 and tyrosine kinase 2 (Tyk2), is an immunomodulatory drug to treat inflammatory diseases including psoriasis (2). In clinical trials, tofacitinib has shown efficacy for plaque psoriasis both as an oral formulation in two phase III trials, and as an ointment formulation in a phase II trial (2). The primary mode of action of tofacitinib has been thought to be the modulation of JAK-STAT signalling in T cells, which is involved in IL-23 signalling, Th17 differentiation and CD4+ T-cell activation. However, in addition to T cells, JAK/STAT signalling is also active in the epidermis of psoriasis lesions (2, 4), and the importance of epidermal JAK/STAT signalling is supported by the psoriasis-like skin phenotype in a mouse model with keratinocyte-specific overexpression of STAT3 (5). Thus, tofacitinib may also regulate cytokine signalling in keratinocytes. The aim of this study was to test the hypothesis that tofacitinib has direct effects on cytokine signalling in keratinocytes.
Human primary keratinocytes from adult skin were obtained from Thermo Fisher Scientific (Stockholm, Sweden) and cultured in Epilife medium (Thermo Fisher Scientific). IL-22 (20 ng/ml, R&D systems, Minneapolis, Minnesota, USA) and tofacitinib (0.6 µM, Pfizer, Stockholm, Sweden) were used to treat primary keratinocytes. Dimethyl sulphoxide was used as a vehicle control for tofacitinib. Four-mm punch biopsies were collected from healthy volunteers (n = 5), non-lesional and lesional skin of patients (n = 6) diagnosed with chronic plaque psoriasis, and epidermal CD45– cells (mainly keratinocytes) were isolated as described previously (6). Peripheral blood mononuclear cells (PBMCs) were isolated from blood, by using Ficoll (GE Health-care, Stockholm, Sweden) density separation. The collection of human samples was performed according to the principles of the Declaration of Helsinki and all subjects provided written informed consent.
Western blot analysis for pSTAT3, STAT3, and β-actin was performed using anti-mouse STAT3 (Santa Cruz Biotechnology, Dallas, Texas, USA), anti-rabbit pSTAT3 (Tyr705 and Ser727) (Cell Signalling Technology, Stockholm, Sweden) and β-actin (Sigma-Aldrich, Stockholm, Sweden).
Total RNA was isolated using phenol chloroform separation from human primary keratinocytes treated with IL-22, with or without pre-treatment with tofacitinib for 6 h. Gene expression profiling was performed using Affymetrix GeneTitan ST 2.1 array (Thermo Fisher Scientific). Microarray data was analysed using Affymetrix standard protocol and fold change, p-value and q-values (p < 0.05, false discovery rate < 10%) were calculated using significance analysis for microarray (SAM), Multiple Experiment Viewer (MEV) (TM4) (microarray data is available on NCBI Gene Expression Omnibus, accession number GSE104509). Differentially expressed genes were used for network analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and gene ontology term enrichment analysis using Cytoscape (ClueGo, Institute for Systems Biology, Seattle, Washington, USA) and online tool EnrichR (Ma’ayan Laboratory, Icahn School of Medicine at Mount Sinai, New York, USA) respectively.
Expression of JAK 1, JAK2, JAK3, Tyk2 (Thermo Fischer Scientific), Egr-1 and S100A7 (Integrated DNA Technologies, Coralville, Iowa, USA) was determined by TaqMan based predesigned qPCR assays. Gene expression was normalized based on housekeeping gene 18S (18S fwd: CGGCTACCACAT CCAAGGAA; rev: GCTGGAATTACCGCGGCT, probe: TGCTGGCACCAGACTTGCC CTC) using ΔCt calculation. Student’s t-test or Mann–Whitney U test was used for statistical analysis of qRT-PCR data. p-values < 0.05 were considered statistically significant.
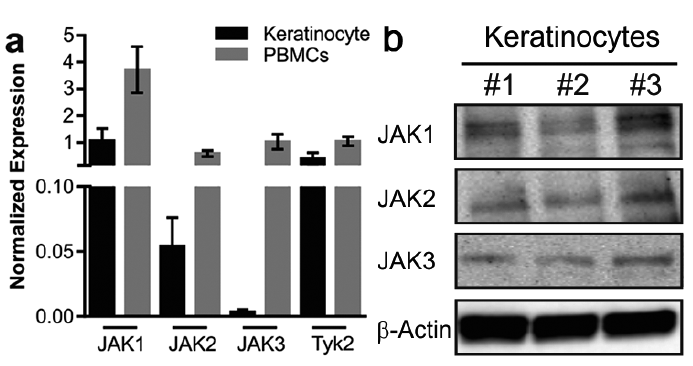
To investigate whether the potential targets of tofacitinib are expressed in keratinocytes, we first examined the expression of JAK1, 2, 3 and Tyk2 in cultured keratinocytes in relation to PBMCs. qRT-PCR analysis demonstrated that all 3 JAKs as well as Tyk2 are expressed in keratinocytes. JAK1 and 2 and Tyk2 were expressed at a comparable level to PBMCs, while JAK3 had lower expression in keratinocytes (Fig. 1a). Furthermore, protein expression of JAK1, 2 and 3 in cultured keratinocytes was demonstrated by Western blot (Fig. 1b).
Fig. 1. The molecular targets of tofacitinib are expressed in keratinocytes. Total RNA was isolated from primary human keratinocytes and peripheral blood mononuclear cells (PBMCs) and the expression of JAK1, 2, 3 and Tyk2 was analysed with quantitative real-time PCR (qRT-PCR) (a). Total protein was isolated from proliferating primary keratinocytes and the expression of JAK1, 2, 3 and β-actin was detected using Western blot (b).
Next, we aimed to investigate whether tofacitinib can modulate cytokine signalling in keratinocytes. To this end, primary human keratinocytes were treated with an activator of JAK-STAT signalling, IL-22 (20 ng/ml), with or without pre-treatment with tofacitinib. qRT-PCR analysis of 2 downstream genes of IL-22, S100A8 and S100A9, showed that tofacitinib suppressed IL-22-induced expression of these genes (Fig. 2). As expected, Western blot analysis revealed that IL-22 induced STAT3 phosphorylation (Tyr705 and Ser727) at 20, 30 and 60 min (Fig. 3a–b). Pre-treatment with tofacitinib prevented IL-22-induced STAT3 phosphorylation (Fig. 3a–b), indicating that tofacitinib has direct effect on keratinocytes. In addition to STAT3, tofacitinib pre-treatment also prevented IL-22-induced phosphorylation of STAT1 (Fig. 3c).
Fig. 2. Tofacitinib rescues expression of IL-22-induced genes S100A8 and S100A9. Human primary keratinocytes were treated with different concentration of tofacitinib (0.1, 0.3 and 0.6 µM) followed by interleukin (IL)-22 treatment (20 ng/ml). Expression of 2 IL-22 downstream genes, S100A8 and S100A9, was measured by quantitative real-time PCR (qRT-PCR). Bars depict mean±standard deviation of experiments performed in triplicate. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (Student’s t-test).
Fig. 3. Tofacitinib prevents IL-22-induced phosphorylation of STAT3 and STAT1. (a–c) Primary human keratinocytes were treated with 20 ng/ml IL-22 for 10, 20, 30 or 60 min 1 h upon treatment with 0.6 µM tofacitinib. Equal amounts of total cell lysates were subjected to SDS–PAGE followed by Western blot analysis using anti-STAT3, anti-phospho-STAT3Tyr705 (a), anti-phospho-STAT3 Ser727 (b), anti-pSTAT1, anti-phospho-STAT1 (c) and anti-β-actin antibodies. Data are representative of 3 independent experiments.
Thereafter, gene expression profiling was performed to determine the effects of tofacitinib on the keratinocyte transcriptome. A total of 898 differentially expressed genes (DEGs) were identified in keratinocytes treated with IL-22 at 6 h. Remarkably, pre-treatment with tofacitinib could prevent the majority of IL-22-mediated gene expression changes, as deregulation of 93% of IL-22-regulated genes was partially prevented by tofacitinib. Out of these DEGs, 193 genes were significantly differentially expressed both between IL-22-treated vs. untreated cells, and between tofacitinib and IL-22 vs. IL-22-treated cells (Fig. 4a, Table SI). Notably, all of the IL-22-regulated genes were also significantly regulated by tofacitinib in the opposite direction (Fig. 4a). To determine if these 193 genes regulated by tofacitinib were clustered into functionally related groups, a gene network analysis was performed. Network analysis showed clustering of JAK-STAT signalling pathway along with regulation of T-cell activation, type I interferon signalling, IL-10 production, epidermal and endothelial cell differentiation (Fig. 4b). KEGG pathway enrichment analysis of the same transcripts demonstrated that genes regulating the JAK-STAT signalling pathway (p < 0.001) along with TNF-signalling (p < 0.001) and cytokine-cytokine receptor interaction (p < 0.05) were prominently altered by tofacitinib (data not shown). Analysis of Gene Ontology (GO) terms (biological processes) identified biological processes associated with psoriasis, including keratinocyte differentiation and response to cytokines (data not shown). These biological functions regulated by tofacitinib mirror the known function of IL-22 in psoriasis, i.e. the regulation of keratinocyte proliferation, inhibiting terminal differentiation of keratinocytes, and inducing the expression of antimicrobial peptides and chemokines (7–9) and suggest that tofacitinib can reverse these effects. Altogether, these results demonstrate that tofacitinib can specifically target the JAK-STAT signalling pathway and regulate psoriasis-related gene networks in keratinocytes, in addition to its previously recognized function to suppress differentiation of pathogenic T-cell subsets.
Fig. 4. Tofacitinib reverses IL-22-induced transcriptomic alterations in keratinocytes. Primary human keratinocytes were treated with IL-22, with (tofacitinib+IL-22) or without (IL-22) pre-treatment with tofacitinib, or medium as a control (ctrl). (a) Heat map showing 193 genes significantly differentially expressed both between IL-22-treated vs. control cells, and between IL-22-treated cells with or without tofacitinib pre-treatment. p < 0.05. (b) Gene Ontology clustering (KEGG) was performed in Cytoscape using the ClueGo plugin on the 193 genes significantly differentially expressed both between IL-22 treated vs. control cells, and between IL-22-treated cells with or without tofacitinib pre-treatment. Each node represents a signalling pathway (Benjamini–Hochberg p value < 0.05), and different colours represent different cellular pathways. The edges reflect the relationships between the terms based on the similarity of their associated genes.
S100A7 and EGR1, two genes that were significantly regulated by tofacitinib were selected for validation. S100A7 is an antimicrobial peptide upregulated in psoriasis, which regulates keratinocyte proliferation and differentiation (10). EGR1 is a transcription factor regulating cell growth, cell differentiation and survival (11). qRT-PCR analysis confirmed the significant upregulation of S100A7 and downregulation of EGR1 by IL-22 in keratinocytes. Tofacitinib reversed the IL-22-mediated regulation of both S100A7 and EGR1 at all studied time-points (Fig. 5a–b). To confirm the relevance of these findings in psoriasis, the expression of S100A7 and EGR1 was analysed by qRT-PCR in sorted CD45– epidermal cells (mainly keratinocytes) from lesional and non-lesional skin of psoriatic patients and healthy controls. In line with our results and previous studies (10), we observed that S100A7 was upregulated in lesional keratinocytes (Fig. 5c). By contrast, the expression of EGR1 was downregulated in the lesional keratinocytes compared with healthy keratinocytes (Fig. 5d). These results further confirm that tofacitinib can partially reverse psoriasis-associated changes in keratinocytes.
Fig. 5. Tofacitinib blocks regulation of S100A7 and EGR1 in keratinocytes. Human primary keratinocytes were pre-treated with tofacitinib one hour prior to interleukin (IL)-22 treatment. The expression of S100A7 (a) and Early Growth Response 1 (EGR1) (b) was analysed by quantitative real-time PCR (qRT-PCR) at the indicated time points after IL-22 treatment (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test). qRT-PCR analysis of (c) S100A7 and (d) EGR1 in sorted epidermal non-immune (CD45–) cells from healthy (H) skin and non-lesional (NL) and lesional (L) skin from psoriasis patients (n = 5 (H), n = 6 (NL and L)). *p < 0.05, **p < 0.01 (Mann–Whitney U test).
This study reveals that the effect of tofacitinib can be more widespread than only targeting T cells, as it can also directly act on keratinocytes. The efficient suppression of JAK-STAT signalling in keratinocytes may contribute to the efficacy of topical tofacitinib treatment in psoriasis and atopic dermatitis, as recently demonstrated in a phase II randomized clinical trials (2, 12). These results are in line with recent findings, showing a rapid reduction in keratinocyte-derived mediators in the skin of patients with psoriasis treated with tofacitinib (13), and support a multifaceted mechanism of action of tofacitinib, with effects on multiple cell types. The direct effect of tofacitinib on keratinocytes can have relevance for systemic as well as for topical treatment with tofacitinib, and possibly also other JAK inhibitors.
The authors would like to thank all patients and control subjects who contributed to this study.
This work was supported by the Inflammation Europe ASPIRE 2014 research award by Pfizer, the Swedish Skin Foundation (Hudfonden), the Swedish Research Council (Vetenskapsrådet) and the Swedish Psoriasis Foundation (Psoriasisfonden). E.S. was supported by the Stockholm County Council and the Swedish Research Council (Vetenskapsrådet). E.S. has received the Inflammation Europe ASPIRE 2014 research award, an independent research grant from Pfizer, for this study. E.S. has participated in advisory boards or panels or given lectures for Novartis, Abbvie, Eli Lilly, UCB and Pfizer Inc.


 Click to show fullsize
Click to show fullsize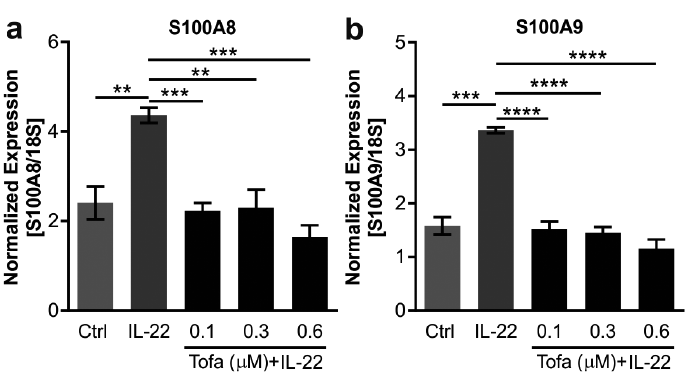 Click to show fullsize
Click to show fullsize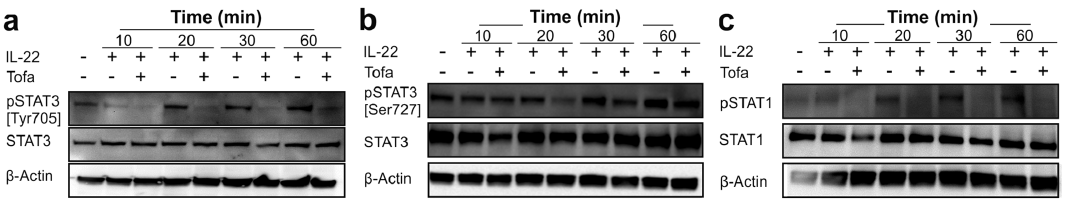 Click to show fullsize
Click to show fullsize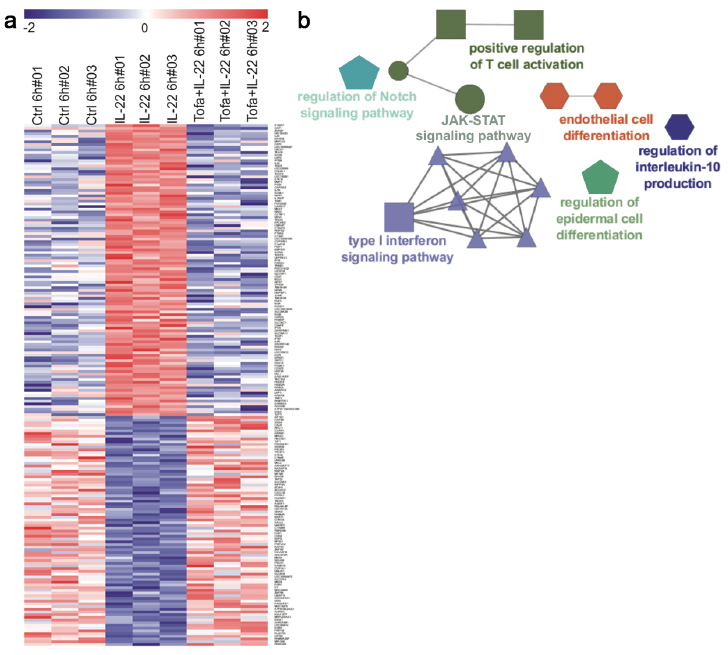 Click to show fullsize
Click to show fullsize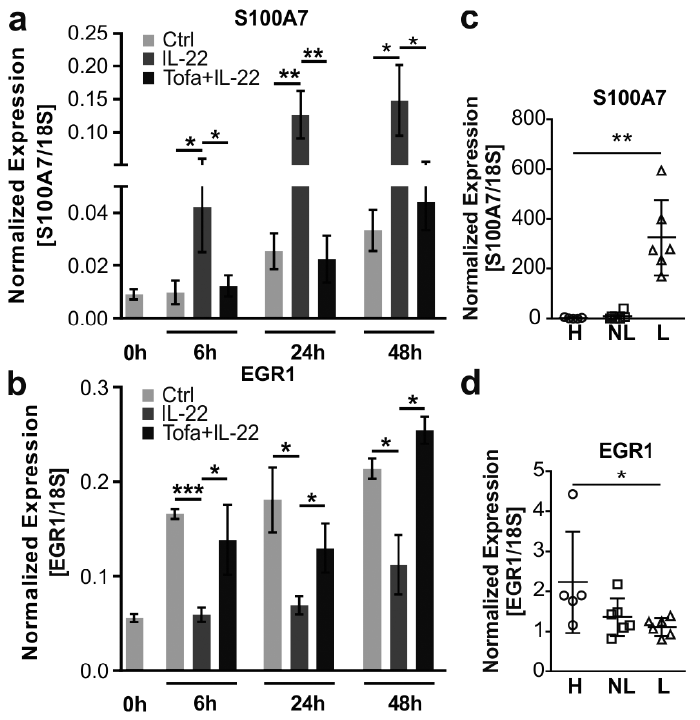 Click to show fullsize
Click to show fullsize