


1Department of Dermatology and Allergy Centre, and 2Department of Clinical Biochemistry and Pharmacology, Odense University Hospital, DK-5000 Odense, Denmark. E-mail: anette.bygum@rsyd.dk
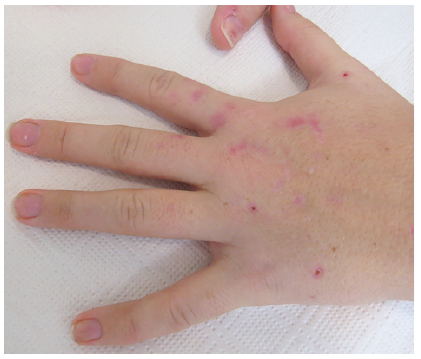
A 17-year-old girl was referred to the department of dermatology due to blisters and sores on both hands for some months. She reported having fragile skin, tense blisters, slow healing sores and development of scars. In addition to the cutaneous symptoms she had muscle weakness in her arms and hands, along with reduced sensibility acrally. Before her referral, a neurological examination confirmed reduced muscle strength in the hands and decreased peripheral sensibility. Magnetic resonance imaging of the cerebrum, and electroneurography, were normal. A subsequent psychiatric evaluation concluded that she was psychologically fragile, but found no evidence of depression or attention deficit hyperactivity disorder. Biochemically she had prolonged raised alanine aminotransferase (ALAT) of 181-106-81-66 U/l [normal 10–45 U/l], while complete blood count, kidney and other liver function tests were normal. The medical treatment at time of presentation was oral contraceptives and a daily multivitamin tablet.
What is your diagnosis? See next page for answer.
Fig. 1. Seventeen-year-old girl with fragile skin, sores and scars on her hands.
Acta Derm Venereol 2018; 98: 815–816.
Diagnosis: Porphyria cutanea tarda (PCT)
Blisters, sores and, especially, vulnerable skin on the patient’s dorsal hands were suggestive of porphyria cutanea tarda (PCT) or pseudoporphyria. A few milia could be seen on her dorsal hands, while she had no hyperpigmentation, hypertrichosis or notable scars on her face. The blistering tendency seemingly spontaneously improved, which could reflect a seasonal variation, as she was seen in December, when sun exposure in Denmark is low.
In PCT phototoxic heme precursors accumulate and lead to blisters and erosions on light-exposed areas (1–5). The healing process is slow and the sores typically heal with scars. Dark urine can also be seen due to excess excretion of porphyrins (2), and this had been noticed earlier by our patient.
Due to her unspecific, but persisting, neurological complaints, with temporary worsening, variegate porphyria and hereditary coproporphyria was also suggested. The possibility of self-inflicted skin lesions was likewise considered.
Biochemical testing was indicative of iron overload: P-ferritin 118 [normal 15–200] µg/l, P-iron 41 [5–18] µmol/l, P-transferrin 25 [25–45] µmol/l, and transferrin saturation 0.81 [0.04–0.40]. Tests for human immunodeficiency virus and hepatitis B and C were negative.
PCT is the most common of the porphyrias. Variegate porphyria and hereditary coproporphyria may show the same cutaneous manifestations as PCT, with blisters, ulcers and scars. To distinguish PCT from the acute porphyrias, it is necessary to investigate the heme intermediate pattern in urine, faeces and plasma (1–3). Porphyrin analysis revealed elevated excretion of, especially, uroporphyrin in her urine, with an uroporphyrin/creatinine ratio of 320.6 × 10–6 [normal 0.0–5.9 × 10–6] and a normal excretion of 5-aminolaevulinic acid and porphobilinogen. Spectrofluorometric scanning of plasma showed an emission peak at 620 nm and faeces analysis showed a F-coproporphyrin III/I ratio < 2. The overall interpretation of the heme excretion pattern was compatible with PCT and there was no biochemical evidence of acute porphyria.
PCT appears in a sporadic form, sPCT, and an inherited form, fPCT, with reduced penetrance (3, 4). Mutations in the UROD gene can be found in 24–53% of patients with PCT (3, 5).
The typical age at onset of sPCT is approximately 40–50 years of age, while fPCT often occur earlier in life, on a mean of 5 years before sPCT (3, 4). Onset in adolescence is quite unusual.
Among the patient’s first-degree relatives there was no family history of porphyria. However, her great grand-mother had experienced a skin disease affecting her hands and had received a diagnosis of porphyria. An investigation for fPCT was performed with genetic testing of the UROD gene encoding the enzyme uroporphyrinogen decarboxylase (UROD) and a mutational analysis of the genes encoding HMBS (~acute intermittent porphyria), CPOX (~hereditary coproporphyria) and PPOX (~varigate porphyria) without abnormal findings.
Considering the young age of the patient and her family history, we were surprised not to find an UROD mutation. This could be due to a mutation in introns, promotor or regulatory regions, or an as-yet unidentified disease-causing mutation outside the UROD locus, which may be involved in reducing UROD enzyme activity, as suggested by Elder (6).
Due to abnormal iron parameters and the known association between hemochromatosis and PCT (1–4), the patient was also tested for mutations in the HFE gene. The result showed that she was homozygous for the mutation p.Cys282Tyr, which is indicative of hereditary hemochromatosis (7, 8).
In patients with iron overload, phlebotomy is an accepted treatment and unnecessary sources of iron should be stopped. The raised ALAT is a typical finding in patients with active PCT, due to a concomitant liver involvement (1, 6). It can also be related to hemochromatosis (7).
The patient was advised to stop all alcohol intake, reduce sun exposure, and consider another contraceptive method, because oestrogen is a known provoking factor of PCT (1–4). In addition, low-dose hydroxychloroquine can be used (1, 3, 6).
An explanation for the neurological symptoms in this patient has still to be found. It is possible that these symptoms may have a psychogenic background or may be related to hemochromatosis (7, 8).


 Click to show fullsize
Click to show fullsize