


1Department of Dermatology & STD, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 2Department of Infectious Disease, Peking University First Hospital, Beijing, 3Biomedical Research Institute, Shenzhen Peking University, The Hong Kong University of Science and Technology Medical Center, Shenzhen, Guangdong Province, 4Department of Dermatology & STD, Affiliated Hospital of North Sichuan Medical College, Nanchong, Sichuan, China
#These authors contributed equally to this article.
Acne vulgaris has been postulated to have a gastrointestinal mechanism; however, little is known about gut microbiota dysfunction in this condition. The aim of this cross-sectional study was to investigate whether the gut microbiota is altered in acne. Faecal bacterial diversity was analysed in 43 patients with acne and 43 controls, using hypervariable tag sequencing of the V3–V4 region of the 16S rDNA gene. Distinct differences were found in microbial diversity between patients with acne and controls (Shannon diversity index (p = 0.009) and Simpson diversity index (p = 0.01)). At the phylum level, the abundance of Firmicutes was lower in the patient group, but that of Bacteroidiain was higher. The most significantly depleted taxa in acne were Clostridia, Clostridiales, Lachnospiraceae and Ruminococcaceae genera, which are potentially beneficial. In conclusion, patients with acne vulgaris have gut microbial dysbiosis; further study is needed to understand its role in the pathogenesis of acne.
Key words: acne vulgaris; gut microbiota; bacterial diversity.
Accepted May 9, 2018; Epub ahead of print May 14, 2018
Acta Derm Venereol 2018; XX: XX–XX.
Corr: Guiqiang Wang, Department of Infectious Disease, Peking University First Hospital, No.8 Xishiku Street, Xicheng District, Beijing, China. E-mail: John131212@126.com; and Xia Xiong, Department of Dermatology & STD, the Affiliated Hospital of Southwest Medical University, No. 25 Taiping Street, Luzhou, Sichuan, China. E-mail: xiongxia789@126.com
This study presents the difference of gut microbes between acne vulgaris patients and healthy controls. Comparing with healthy controls, acne patients show the decreased diversity of gut microbiota and the increased ratio of Bacteroidetes to Firmicutes which has been reported to be the enterotype of the Western diet. These data validate the role of western diet in promoting the development of acne vulgaris and suggest the diet and probiotic-based therapy in preventing and treating acne vulgaris.
Acne vulgaris is a chronic inflammatory skin disease of the pilosebaceous units that is estimated to affect up to 80–90% of adolescent patients, particularly in developed nations (1). In general, the pathogenesis of acne is characterized by increased production of sebum, inflammatory mediators in the skin, and follicular hyperkeratinisation of pilosebaceous ducts. The role of Propionibacterium acnes is not clear, as this organism is ubiquitous in healthy adult human skin. However, an altered phenotype of P. acnes in acne lesions may be more associated with the disease and may also be pro-inflammatory (2, 3). Although genetic predisposition can strongly influence the risk of developing acne, the role of environmental factors, especially the “Western style” diet, in the pathogenesis of acne has been suggested (4, 5). The “Western style” diet includes milk, other dairy products, refined carbohydrates, chocolate, and saturated fats, which contribute to the formation of acne by activating nutrient-derived metabolic signals (6, 7). Dietary supplementation with omega-3 fatty acids has been reported to help decrease lesions in patients with mild to moderate acne (8). The ratio of omega-6 to omega-3 fatty acids is higher in “Western style” than in non-Western diets (9).
Accumulating evidence indicates that the gut microbiota associated with the “Western style” diet play a significant role in the development of a number of disease states, such as immune, metabolic, and even skin, diseases (10–12). High-fat diets have been reported to reduce the levels of gut microbes and increase the concentration of lipopolysaccharides (LPS), which contribute to the development of systemic inflammation by impairing colonic epithelial integrity and barrier func-tion, decreasing mucus layer thickness and increasing the secretion of pro-inflammatory cytokines (13–15). However, there is no direct evidence that imbalance of gut microbiota contributes to the pathogenesis of acne vulgaris.
It can be postulated that acne vulgaris features a gastrointestinal mechanism based on the theory of microbiota-gut-brain axis (16). The latest research has shown that communication between the gut and brain plays a critical role in maintaining optimal health, and that changes in gut microbiota may affect central nervous system function and neuroendocrine responses, such as the hypothalamus pituitary adrenal axis (HAP) (16–18). HAP modulates most of a number of pathways and hormones implicated in the pathogenesis of acne, such as androgens, insulin-like growth factor, corticotrophin-releasing hormone, ectopeptidases, adrenocorticotropic hormone, and glucocorticoids (19). Both inflammation and the innate immune system play a central role in the pathogenesis of acne (20). Gut microbiota exert effects on the brain through the immune system, and the balance of gut microbiota may alter regulation of the inflammatory response (16). However, it is not known whether there is dysbiosis of the gut microbiota underlying acne vulgaris. Also, there are no data on the gut microbiome in similar skin diseases, e.g. rosacea, where antibiotics may be used long term. This study was conducted to analyse the faecal microbiota composition of patients with acne vulgaris and to compare it with that of healthy controls.
A total of 43 patients diagnosed with acne vulgaris and 43 age- and sex-matched healthy controls were enrolled in this study. All participants were students from middle schools in Luzhou city, Sichuan province, and the Southwest Medical University between August 2016 and May 2017. The severity of acne vulgaris was judged by the number of inflammatory lesions on half of the face, according to the Japanese Acne Study Group criteria (21). The severity of mild, moderate, severe and very severe acne were defined as S1, S2, S3 and S4. Patients and controls were asked precise questions regarding their medical history, as well as any treatment they had received. All the patients included in this study were treatment-naïve, never having received any oral medicine for acne, such as antibiotics, isotretinoin, herbs, etc. Although there are no published reports on the effect of isotretinoin on the gut microbiome, isotretinoin treatment history should be considered as an exclusion criteria, because it has been considered the first-line clinical medicine for acne, and may intervene in disease stage and pathological factors associated with the disease. In addition, all patients were asked questions regarding any treatment they had received for other diseases. Only those who had not used antibiotics, glucocorticoids, immunosuppressive drugs, or herbs for treating other diseases within 6 months were included, according to previous studies, which considered 3–6 months as the cut-off time of treatment history (22–24). The exclusion criteria were the presence of other dermatosis, such as contact dermatitis, atopic dermatitis, eczema, psoriasis, ichthyosis, cutaneous fungal infection, an immunodeficiency or any other immunological disorder, metabolic diseases (e.g. obesity, fatty liver disease, diabetes mellitus), malignancy, any other serious internal disease, smoking, and alcohol abuse. Pregnant and nursing women, people with a known history of gastrointestinal tract surgery (e.g. gastrectomy, bariatric surgery, or colectomy) were also excluded. Clinical data, including age, sex, and body mass index (BMI), were recorded at the time of sample acquisition. All patients and controls were also questioned about their dietary habits over the past 6 months, such as consumption of spicy food, dairy products, fried and fatty food, refined carbohydrates, vegetables, and fruit. The frequency of consumption of each kind of food listed above was evaluated as follows: frequently (consuming special food every 3 days or less than 3 days), sometimes (once a week and more than 3 days), occasionally (once a month and more than once a week), none (more than 1 month). All participants gave written informed consent for the use of their data and samples for scientific purposes, and the study was approved by the ethics committees of the hospital affiliated to Southwest Medical University.
Fresh stool samples from each person were collected in sterile containers, immediately homogenized and divided into 10 aliquots of 220 mg, then frozen at –80°C within 30 min. During the period from defecation to division, samples were kept at 4°C. Microbial DNA was extracted from the faecal material of each sample using the QIAamp DNA Stool Minikit (Qiagen Ltd, Strasse, Germany) following the manufacturer’s instruction. Partial 16S rDNA gene sequences were amplified from each extracted DNA using the primer pair, F341F (5’-ACTCCTACGGGRSGCAGCAG-3’) and R806R (5’- GGACTACVVGGGTATCTAATC-3’), which targets the V3–V4 region of the 16S rDNA gene sequence. KAPA HiFi Hotstart ReadyMix PCR kit was used for high-fidelity amplification. The quality of amplicons was assessed using NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and 2% agarose gel electrophoresis. The 16S rDNA gene amplification products were sequenced in Illumina Hiseq 2500 instrument (Illumina, San Diego, CA, USA) with 2×250 base pair (bp) paired-end (PE) sequencing, at Realbio Technology (Shanghai, China).
PANDAseq was applied to assemble overlapping paired-end reads. Quality control retained sequence had lengths between 220 and 500 nt, a mean sequence quality score of > 20, and reads with N bases < 3. Sequences of 16S rDNA were grouped into operational taxonomic units (OTUs) using the mean neighbour algorithm. To calculate downstream diversity measures (α and β diversity analysis), sequences with a distance-based similarity of ≥ 97% were assigned to the same OUT, using USEARCH after removal of singletons. Furthermore, chimeric sequences were filtered using ChimeraSlayer. Subsampling of 22,934 reads was conducted in each sample, while sequencing at sufficient depth. Bacterial taxonomy assignment was performed by using the RDP database and classifier (RDP, http://rdp.cme.msu.edu).
Rarefaction curves were generated with QIIME to test the current sequencing depth. Shannon and Simpson diversity indices were used to estimate the α diversity, which represented the species abundance in a single sample and were compared between groups by Wilcoxon rank sum test and Kruskal-Wallis rank sum test using R3.1.0. The communities were compared based on phylogenetic distances using the weighted UniFrac metric to represent β diversity. Principal coordinates analysis (PCoA) was performed on the resulting matrix of distances between each pair of samples using R3.1.0. MRPP analysis. Heatmap was also employed. Based on the UniFrac phylogenetic distance, significant test for the clustering of samples in the study was carried out by one-way analysis of similarities (ANOSIM). LefSe analysis was applied to identify different abundant bacterial taxa among groups. Only those taxa that obtained a log linear discriminant analysis (LDA) score >2 were ultimately considered.
Statistical analysis of gut microbial differences between acne samples (AS) and control samples (CS) was performed by Wilcoxon rank sum test and Kruskal-Wallis rank sum test using R3.1.0. All p-values reported are two-sided, and p <0.05 was considered statistically significant. We also applied the Benjamini and Hochberg false discovery rate test (FDR) or calculated the 95% confidence intervals (CI) if the FDR q value was > 0.1.
PICRUSt provided a number of scripts that could be useful for analysing both 16S rRNA gene relative abundances and the predicted metabolic data. The protein sequences of genes in the merged gene catalogue were aligned to the Kyoto Encyclopedia of Genes and Gemos (KEGG) bioinformatics database (8th KEGG release, December 2014). The data were analysed statistically using STAMP, which allows data filtering and the application of different statistical tests.
This study included 43 patients with acne and 43 age- and sex-matched healthy controls. The background of patients and controls are shown in Table I. There were 26 males in each group. Age ranged from 14 to 25 years. Patients tended to consume dairy products, fried and fatty food more frequently than did healthy controls. All persons had a BMI less than 24 kg/m2. In the patient group, there were 10 cases with very severe acne, and 12, 12 and 9 cases showed mild, moderate and severe acne, respectively. Mean disease duration was 37.32 ± 7.84 months. During the study, no patients were withdrawn, and there were no missing data. In addition, all the faecal samples in the study met the standard of analysis.
Table I. Background of patients with acne and healthy controls
A total of 86 faecal samples were obtained from the study population for sequencing. After processing with PANDAseq, a total of 3,989,761 high quality reads with a mean of 46,393 ± 10,876 reads per sample were obtained. The 86 samples clustered a total of 708 OTUs at a 3% dissimilarity cut-off, with 86–275 OTUs per sample.
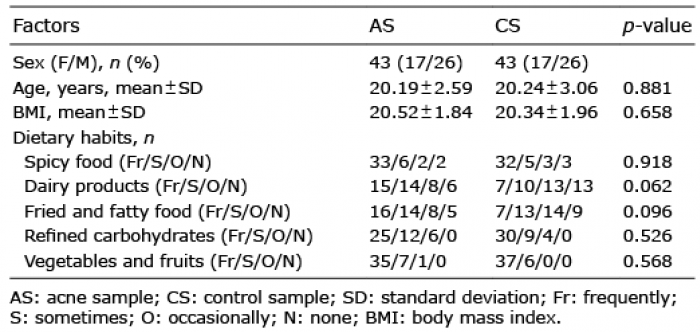
Compared with CS, microbial diversity was significantly decreased in AS, as calculated by the Shannon diversity index (p = 0.009) and Simpson diversity index (p=0.01) (Fig. 1a, b). However, no significant difference in microbial diversity was observed among patient subgroups divided by disease severity (Fig. 1c, d). By comparing the OTUs between acne and healthy samples, we found that most OTUs were shared by different samples (Fig. 2a). PCoA were implemented to assess discrepancies based on OTUs with different relative abundances. Based on the PCoA plot, we found that there was a tendency to form 2 clusters, a tight acne cluster (Fig. 2b, blue dots) and a disperse healthy cluster (Fig. 2b, red dots). To investigate the different structures of gut microbes between groups, we calculated UniFrac phylogenetic distances of the microbe composition among subjects. ANOSIM analysis indicated the significance of clustering samples into acne and healthy groups (R=0.046, p = 0.029) (Fig. 2c). MRPP analysis indicated a significant difference between healthy and acne samples (p = 0.034, data not shown). Heatmap was also applied to evaluate the similarity of each sample composition, which showed a trend of difference between the 2 subject groups (Fig. 2d). There was no significant difference observed in the dissimilarity of bacterial structures among patient subgroups with different severity (Fig. S1).
Fig. 1. Comparisons of different diversity indices in acne samples and healthy samples. Simpson and Shannon diversity index, representing community richness, were calculated for the acne samples (AS) and the control samples (CS). (a) Simpson diversity index (p = 0.01) between AS and CS. (b) Shannon diversity index (p = 0.009) between AS and CS. (c) Simpson diversity index (p = 0.466) among acne groups with different severity. (d) Shannon diversity index (p = 0.374) among acne groups with different severity.
Fig. 2. Comparison of the gut microbiome between acne and control samples. (a) Venn analysis of bacterial operational taxonomic units’ (OTUs) composition between acne samples (AS) and the control samples (CS). (b) Principal coordinates analysis (PCoA) plot with different relative abundances of OTUs between AS and CS. (c) Difference between and within groups of AS and CS were assessed by one-way analysis of similarities (ANOSIM) analysis. (d) Heatmap with weighted UniFrac phylogenetic distances of the microbial taxa among subjects.
A total of 242 bacterial taxa were analysed in our samples, of which 126 taxa were at the genus level. To further investigate which taxa were distinct among groups, LefSe analysis was applied. We found 38 differentially abundant taxa between the AS and CS groups, all of which had a log LDA score > 2. Results are presented in Fig. 3a, with blue and red colours indicating a decrease and increase of abundance in AS, respectively. Then, Wilcoxon rank sum test and Kruskal-Wallis rank sum test were used to perform differential abundance analyses at the phylum, family, class, order, and genus levels (Table II). Normally, bacterial taxa with a p-value < 0.05 and FDR q value < 0.1 were considered to be significantly different. Although all of the FDR q values were > 0.1, the mean abundance of taxa between the 2 groups were different (p < 0.05), and all of the 95% CIs of mean abundance difference of those taxa never spanned 0. Consequently, the 38 bacterial taxa above were considered to be different between AS and CS, of which 19 taxa were at the genus level.
Fig. 3. Bacterial taxa differences between patients with acne and healthy controls. Bacterial taxa significantly enriched in control samples (CS) compared with acne samples (AS) were detected by LefSe (p < 0.05, linear discriminant analysis (LDA)>2 logs). (a) Except 3 taxa, 35 ones were found to be enriched in CS (red bars) compared with AS (blue bars). (b) Box plots with relative abundance of the top 20 differently microbial taxa. (c) Box plots with relative abundance of the different microbial taxa at the general level.
Table II. Relative abundance of acne samples (AS) and healthy samples (CS). Only p-values < 0.05 are shown
Fig. 3b shows the top 20 different abundance of microbes between AS and CS. At the phylum level, the abundance of Firmicutes was lower, but Bacteroidetes was higher in patients with acne. The increased ratio of Bacteroidetes to Firmicutes has been reported to be the enterotype of the Western diet and is associated with some other inflammatory disorders (25–27). Among the 38 differential taxa, only the phylum Bacteroidetes, the class Bacteroidia and order Bacteroidales were more abundant in patients with acne. The members of Bacteroidetes have been reported to be associated with immunity and metabolic processes (28). In another 35 decreased microbes in patients with acne, the most significantly altered taxa were Clostridia, Clostridiales, Lachnospiraceae, Ruminococcaceae, which are potentially beneficial (28, 29).
The 19 different taxa at the genera level in acne samples are shown in Fig. 3c, most of which belonged to Firmicutes phylum. We could separate the patients with acne from the healthy people using the relative abundance of all 38 differential taxa or 19 taxa at the genera level, respectively (Fig. 4a, b).
Fig. 4. Principle component analyses plot of different microbial taxa between acne and healthy samples. (a) PCA plot of 38 different taxa between acne samples (AS) and control samples (CS). (b) PCA plot of 19 different genera between AS and CS.
To explore the functional feature of the gut microbiota in patients with acne, we annotated the gene catalogue by KEGG. At the pathway level, we identified 10 and 23 differently abundant pathways at 2 and 3 levels, respectively, indicating a diverse change in the functions of the acne microbiota compared with controls (Fig. 5a, b). Two categories, including membrane transport and glycan biosynthesis and metabolism exhibited the most significant differences. In the membrane transport, the main decreased pathways in patients with acne were transporters and ABC transporters. Within the glycan biosynthesis and metabolism, relatively enriched pathways were LPS biosynthesis, LPS biosynthesis protein, glycosphingolipid biosynthesis ganglio series, glycosphingolipid biosynthesis globo series, and glycosaminoglycan degradation.
Fig. 5. Distribution of Kyoto Encyclopedia of Genes and Gemos (KEGG) functional categories of KEGG Orthologs (KO) markers. (a) Comparison between the healthy people-enriched and acne patients-enriched markers on level 2 of KEGG functional category. (b) Comparison between the healthy people-enriched and acne patients-enriched markers on level 3 of KEGG functional category. LDA: linear discriminant analysis.
This study demonstrates that the composition of gastrointestinal microbes in patients with acne differs significantly from that in healthy controls. Decreased diversity of gut microbiota was found in the samples from patients with acne, which also occurs in other inflammatory skin diseases, such as Behçet’s disease and psoriasis (23, 29). Previous studies have shown that individuals with a low bacterial richness are characterized by a more pronounced inflammatory phenotype (15).
Extensive research on gut microbiota has shown that long-term dietary trends are linked with features of microbiota structures (25). Phyla positively associated with fat, but negatively associated with fibre, were predominantly Bacteroidetes and Actinobacteria, whereas Firmicutes and Proteobacteria showed the opposite association (22). Dietary interventions, such as including whole-grains in the diet, have been reported to increase the Firmicutes/Bacteroidetes ratio (30). Compared with healthy controls, patients with acne had a lower abundance Firmicutes, but increased Bacteroidetes, in this study, which is consistent with the enterotype of the Western diet. In addition, gut microbiota exert effects on the immune system, and the balance of gut microbiota may change the regulation of inflammatory response (16). Following the communication of bacteria with the local mucosal immune system and intestinal epithelium, cytokines, such as transforming growth factor (TGF), tumour necrosis factor (TNF), interleukin (IL)-6, and IL-10, are produced (16). Inflammation and the innate immune system play a central role in the pathogenesis of acne (20). Inflammatory markers, such as IL-1, TNF-α, IL-6, and IL-8, were present in the early microcomedones and inflamed lesions (31), suggesting that disorder of gut microbiota caused by Western dietary habits could contribute to the early inflammation in acne. In this study, no significant difference was observed in the dissimilarity of bacterial structures among patient subgroups with different severity, which indicated that the disorder of gut microbiota may occur before the clinical manifestation. In fact, a similar dysbiosis between Firmicutes and Bacteroidetes in the human gut has been described in previous studies in association with other inflammatory disorders, such as Crohn’s disease and systemic lupus erythematosus (26, 27).
In this study, besides the disorder of the Firmicutes and Bacteroidetes phyla, the most significantly decreased microbes were Clostridia, Clostridiales, Lachnospiraceae, and Ruminococcaceae, which also have been reported to be less abundant in other inflammatory diseases, such as inflammatory bowel disease (IBD), Behçet’s disease, Crohn’s disease and psoriasis (23, 32–37). Functional analysis based on KEGG suggested that gut microbiota in patients with acne were enriched in the digestive disease pathway. This discovery was consistent with gastrointestinal symptoms of patients with acne. Depletion of Lachnospiraceae and Ruminococcaceae has been reported to be associated with Clostridium difficile infections and nosocomial diarrhoea (32). In addition, Clostridia is a producer of butyrate and contributes to the inhibition of inflammation (29).
A functional analysis using the data obtained from the KEGG showed the LPS biosynthesis pathways were markedly over-represented in the microbiota of AS. Bacteroidetes have been shown to be the main contributors of LPS biosynthesis (38). In 1983, a study involving 80 patients with acne reported that LPS endotoxins from Escherichia coli were present in the serum of patients with acne (39). In this study, there was a trend towards increased E. coli in acne samples (mean 1.22 × 10–2) compared with those of healthy controls (mean 9.2 × 10–3) (p = 0.086), data was not shown. However, whether and how the LPS produced by increased Bacteroidetes or by E. coli contributed to the onset of acne vulgaris, are still unclear and need further confirmation. In addition, glycerolipid and glycerophospholipid metabolism pathways declined slightly in AS, providing an explanation for the high serum cholesterol and lipid levels in patients with acne (15).
The ATP-binding cassette (ABC) transporter, which is widespread in bacteria, was previously thought to be associated with nutrient uptake in bacteria, and later recognized to be involved in diverse biochemical and physiological processes (40). Compared with patients with a high BMI, patients with a low BMI were found to have a significant abundance of several ABC transporter proteins, which was also associated with a high abundance of Clostridium, Blautia, Ruminococcus and Faecalibacterium within the Firmicutes phylum (41). In our study, ABC transporter was significantly decreased in the microbiota of AS. This could be the result of less diversity and a lower abundance of the Firmicutes phylum and its special genera, such as Clostridia, Blautia and Ruminococcus. We speculate that a disorder of gut microbiota leads to an abnormal concentration of ABC transporter subfamily members, which contributes to the pathogenesis of acne. However, this hypothesis needs further investigation.
This study has some limitations. First, the Chinese diet differs markedly from the Western diet. Although the Western diet is postulated to be responsible for the recent increase in acne, the subjects included in this study were not representative of a “classical” Western population. Thus, different results might be obtained if the study participants are classic Westerners. Nonetheless, this study recorded the dietary habits of all participants and found that patients with acne tended to consume dairy products and fried and fatty foods more frequently than did healthy controls. Overgrowth of antibiotic-resistant variants often leads to a loss of diversity, but the participants in this study were not checked. It should be noted, however, that none of the study participants had taken antibiotics for their acne and most of the patients with acne were treatment-naïve. Consequently, there would be fewer antibiotic-resistant variants. The third limitation was that there was only one sequencing measurement sample for each patient. For gut microbiota analysis, most studies shared similar methods of sample collection and detection, and sequencing of 16S rDNA was always performed on one sample per patient (23, 38). In order to reduce variation between samples and the consequently inaccurate result, matters such as fully homogenized stool samples, large sample size, and multiplicity adjustment for analysis have been taken into consideration in this study. Finally, the study only described the phenomenon of disordered gut microbiota in patients with acne. The pathogenesis of microbiota underlining the disease needs further study.
In conclusion, this study explored the differences in gut microbial composition and function between patients with acne and healthy controls. The decreased diversity and Firmicutes/Bacteroidetes ratio in patients with acne is consistent with the enterotype of the Western diet. Several obviously changed microbes and functional pathways were identified that have also been reported to be abnormal in other inflammatory disease, especially IBD, psoriasis, and Crohn’s disease, among others. These findings indicate that the disorder of gut microbiota caused by Western dietary habits could be associated with the development of acne. However, the mechanisms by which the gut microbiome exerts its effects, and the links between the gut microbes and the pathogenesis of skin disease are not yet clear. The bioactive metabolites formed by the interaction of bacterial taxa with dietary components and metabolites are the signalling link from the gut microbiota to the host. Further research will conduct metabolomics analysis on faecal samples from patients with acne and age- and sex-matched healthy controls. In addition, measurement of serum hormones; for example, insulin-like growth factor, will be performed. Further research into the association of gut microbiota, microbial metabolism, and serum hormones will help elucidate the role of the microbiota-gut-brain axis in the pathogenesis of acne.


 Click to show fullsize
Click to show fullsize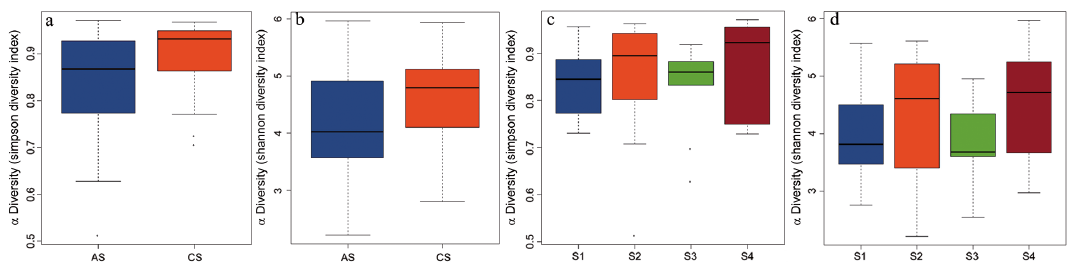 Click to show fullsize
Click to show fullsize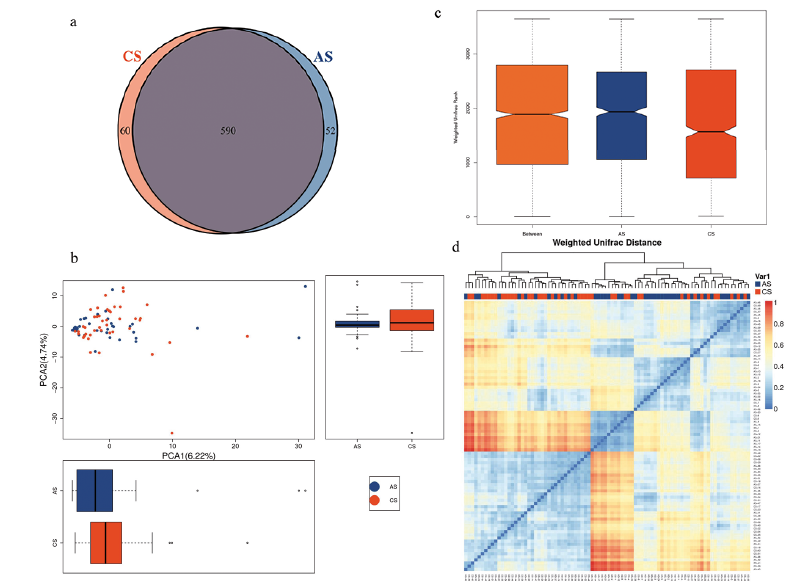 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize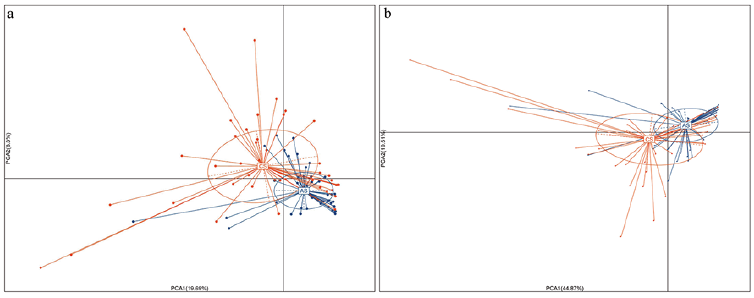 Click to show fullsize
Click to show fullsize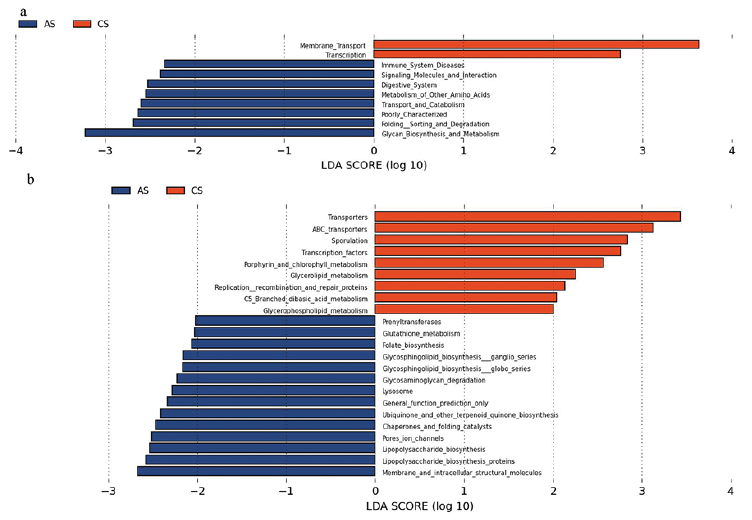 Click to show fullsize
Click to show fullsize