


1Department of Dermatology, 2Department of Pathology, CHRU Tours, FR-37044 Tours, and 3PRES Centre, Val de Loire University, University of Tours, Tours, France. *E-mail: drhcornillier@gmail.com
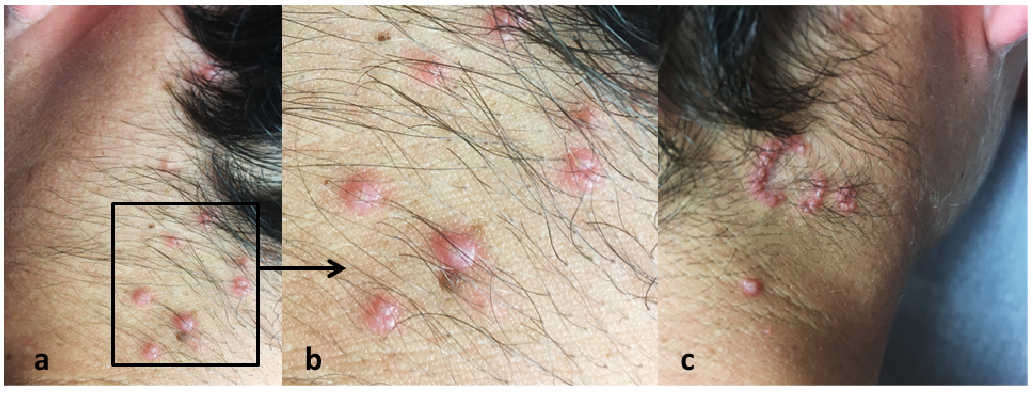
A 14-year-old boy presented with an 8-month history of asymptomatic papules on his neck. The lesions first appeared on the left side and spread to the right side. The patient’s personal and family medical histories were unremarkable, and he was not taking any medication. Clinical examination revealed erythematous papules with umbilicated and keratotic centres, arranged in linear and annular patterns on the neck (Fig. 1).
No other lesions were noted on skin examination and the clinical examination was otherwise normal. Complete blood count, C-reactive protein level, and renal and liver function test results were within normal limits. A skin biopsy was performed (Fig. 2).
What is your diagnosis? See next page for answer.
Fig. 1. (a, b) Erythematous papules with a keratotic centre on the left side of the neck. (c) Papules grouped in an annular pattern on the right side of the neck.
Fig. 2. (a) Intra-epidermal pseudo-cavity connected to the surface, filled with elastin and collagen fibres. Note the thickened epidermis and epithelial mild dermal inflammatory cells infiltrate (hematin-phloxin-saffron, ×200). (b) Higher magnification showing fragmented elastin fibres filling the intraepidermal cavity (orcein ×400).
Acta Derm Venereol 2018; XX: XX–XX.
Diagnosis: Elastosis perforans serpiginosa (EPS)
A diagnosis of elastosis perforans serpiginosa (EPS) may be suspected if skin examination reveals keratotic papules grouped in an annular pattern (1, 2). Differential diagnoses usually includes folliculitis, prurigo nodularis and lichen planus (1). Dermoscopy may be helpful in differentiating EPS from granuloma annulare (3). Histopathological examination revealed a thickened epidermis and a transepithelial channel filled with basophilic material (Fig. 2a), consisting of degenerated keratinocytes, inflammatory cells and elastic fibres (Fig. 2b). Perforating dermatoses are characterized by a transepidermal elimination of various materials including fibres of collagen, elastin or cell debris. EPS is 1 of the 4 primary acquired perforating dermatoses; the other 3 are reactive perforating collagenosis, perforating folliculitis and Kyrle disease (1, 2).
The prevalence of EPS is unknown; only a few cases have been described in the literature. It appears to be more frequent in young males aged 10–30 years. Three subtypes are described: reactive, drug-induced and idiopathic. Reactive EPS is associated with genodermatoses and connective tissue diseases, such as Down, Ehlers-Danlos, Marfan, and Rothmund-Thomson syndromes; osteogenesis imperfecta; morphea; progeria; cutis laxa; and pseudoxanthoma elasticum (1, 2, 4). Drug-induced EPS can be caused by D-penicillamine treatment of Wilson’s disease (5). So-called idiopathic EPS may have a genetic basis because familial occurrence with autosomal dominant inheritance has been described; however, no gene has been identified (6, 7).
EPS lesions can resolve spontaneously after a few months or years, sometimes leaving atrophic or hypopigmented scars. Several therapies have been proposed: topical or intralesional corticosteroids, topical retinoids, oral isotretinoin, imiquimod, cryotherapy, CO2 laser therapy or photodynamic therapy (1, 8, 9). There are currently no specific recommendations for EPS treatment, probably because of the small number of reported cases, the spontaneous improvement, and a lack of randomized controlled studies. No therapy was initially proposed, as the patient did not report functional or aesthetic discomfort. However, when the eruption progressed and became pruritic, treatment with topical retinoids was initiated (tazarotene 0.1% once/day) (10). Partial remission was noted after 3 and 6 months.


 Click to show fullsize
Click to show fullsize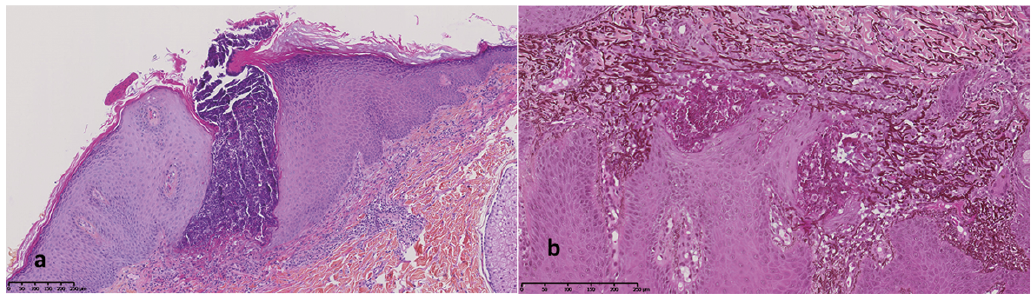 Click to show fullsize
Click to show fullsize