


Department of Dermatology, Nara Medical University School of Medicine, 840 Shijo, Kashihara, Nara 634-8522, Japan. E-mail: fumim@naramed-u.ac.jp
Accepted May 22, 2018; Epub ahead of print May 24, 2018
Lymphomatoid papulosis (LyP) is a cutaneous CD30+ lymphoproliferative disorder (LPD) characterized by chronic recurrent, self-healing papulonodular eruptions and the histological features of malignant lymphoma (1). Patients with LyP are at increased risk of developing lymphoma, particularly mycosis fungoides (MF) and anaplastic large cell lymphoma (ALCL) (1). We describe here an unusual case, in which LyP occurred in a patient with diffuse large B-cell lymphoma (DLBCL) and primary cutaneous ALCL (pcALCL). No cases of LyP associated with both pcALCL and DLBCL have been reported previously (according to a PubMed search using the terms “lymphomatoid papulosis”, “diffuse large B-cell lymphoma”, and “cutaneous anaplastic large cell lymphoma”).
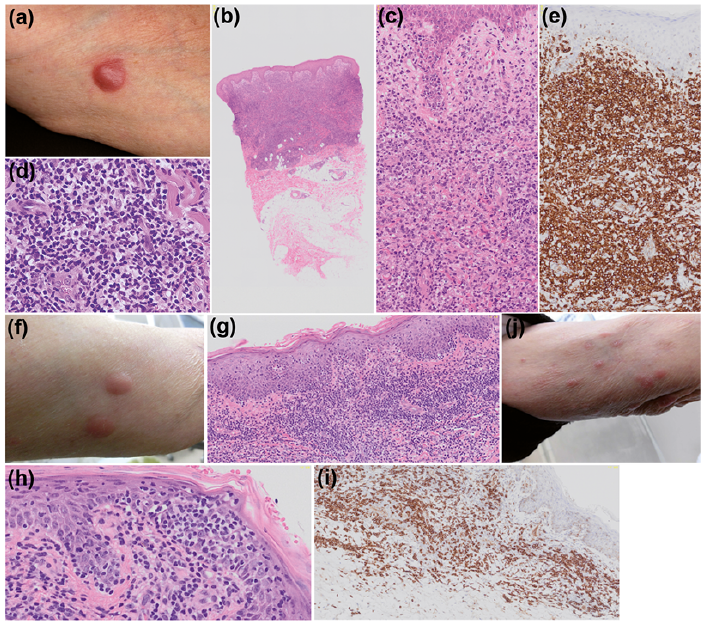
A 79-year-old man presented with a 1.5-cm red tumour on his left arm, which grew over a 1-month period (Fig. 1a). Multiple swollen lymph nodes (LN) were seen on computed tomography (CT) of the chest 16 months previously, and a cervical LN biopsy was performed. A histopathological examination of the LN revealed large atypical cells, which expressed CD20 and immunoglobulin kappa mRNA, surrounded by histiocytes and lymphocytes. He had been diagnosed with T-cell/histiocyte-rich large B-cell lymphoma, a type of DLBCL, and had just received 8 courses of rituximab, which resulted in remission. His medical history included inter-stitial pneumonitis, chronic hepatitis C, hypertension, and chronic kidney disease. A biopsy of the tumour revealed diffuse infiltrates of large pleomorphic lymphoid cells from the dermis to the subcutis (Fig. 1b–d). Immunohistochemistry showed that most tumour cells expressed CD3, CD4 and CD30 (Fig. 1e). The tumour cells were negative for CD20, CD79a, CD10, CD138, the kappa and lambda light chains, Epstein-Barr virus-encoded small RNA, CD56 and anaplastic lymphoma kinase. After a biopsy examination, the tumour partially receded, and the remaining tumour was excised one month later. PCR-based analysis of the excised tumour detected clonally rearranged T-cell receptor β- and γ-chain genes. A diagnosis of pcALCL was made. Shortly after the tumour was excised, the patient noticed that 3 new tumours had developed. On examination, he had 2 tumours on his left arm (Fig. 1f) and 1 on his right knee, all of which measured 1.0×0.8 cm in diameter. A month later, he developed 3 more nodules on his left arm. Six tumours/nodules were excised. Histopathological examination of the tumours showed the epidermotropic infiltration of small to medium-sized lymphocytes with atypical nuclei as well as a Pautrier’s microabscess, consistent with type B LyP (Fig. 1g, h). Most tumour cells exhibited the CD3+CD4+CD30+ phenotype (Fig. 1i). From 2 months after the second surgical procedure, multiple nodules <1.0 cm in size continued to appear every few months for 1 year. These nodules were always restricted to the left arm and disappeared spontaneously within 1 month (Fig. 1j). Their clinical features were consistent with LyP. During this period, the patient developed bilateral axillary lymphadenopathy, and the recurrence of DLBCL was confirmed by a lymph node biopsy examination. He received 5 courses of R-CVP therapy (rituximab with cyclophosphamide, vincristine and prednisone) and 2 courses of rituximab for DLBCL and went into complete remission. However, he died from pneumonia 3 months later. No recurrence of the skin lesions was seen during the last 3 months of the patient’s life.
Fig. 1. (a) Tumour on the left upper arm. (b) Histopathological examination demonstrated diffuse cohesive dermal infiltrates of large pleomorphic lymphoid cells (haematoxylin and eosin staining; ×20). (c) Higher magnification of (b) (×200). (d) Higher magnification of (b) (×400). High-power image of atypical T-cells is shown. (e) Immunohistochemistry detected diffuse positivity for CD30 (×200). (f) Two nodules on the left upper arm. (g) Histopathological examination demonstrated epidermotropic infiltrates of small to medium-sized lymphocytes with atypical nuclei. A Pautrier’s microabscess was detected (haematoxylin and eosin staining; ×100). (h) Higher magnification of (g) (×400). (i) Immunohistochemistry demonstrated that the tumour cells were positive CD30 (×100). (j) Clusters of recurrent, self-healing, nodules on the left upper arm.
Of patients with LyP, 10–60% have associated lymphomas (1–4). These lymphomas include MF, systemic or cutaneous ALCL, and Hodgkin’s lymphoma and can occur before, concurrent with, or after the manifestation of LyP (1–3). Previous studies demonstrated that, in addition to these 3 types of lymphoma, LyP was also associated with other haematological malignancies, including chronic lymphatic leukaemia, acute myeloid leukaemia, B-cell lymphoma, and multiple myeloma, although such cases are rare (4, 5). Non-haematological cancers, including basal cell carcinoma, squamous cell carcinoma and melanoma, have also been observed in patients with LyP (4). Our patient had DLBCL and pcALCL before developing LyP. However, it is possible that the solitary 1.5-cm tumour observed at the initial presentation was caused by LyP type C rather than pcALCL. Since non-regressing lesions >10 mm in diameter are defined as pcALCL, and the tumour’s histological features were compatible with ALCL, the tumour was diagnosed as pcALCL. The multiple nodules/tumours that developed after the solitary tumour were probably LyP lesions because they were small in size, regressed spontaneously, and most of them were restricted to the same site as the first solitary tumour. If the solitary tumour had simply been a larger LyP lesion, a diagnosis of LyP mixed subtype (type B + C) could have been made. However, no cases of LyP associated with DLBCL have been reported.
It has been demonstrated that there is a small subset of patients in whom LyP and pcALCL cannot be distinguished. LyP presents with grouped or disseminated papules and smaller nodules, which regress spontaneously within a few weeks (1). LyP has a wide spectrum of histological manifestations and is classified into 5 histological subtypes (A–E). The histological features of LyP type C and pcALCL are identical (1). pcALCL manifests as a solitary tumour or grouped firm nodules, and spontaneous tumour regression is reported to occur in 10–42% of cases (1, 2). In cases in which it is difficult to determine whether a tumour is a larger LyP lesion or an early-stage pcALCL lesion, a tumour size of more than 2 cm and a lack of spontaneous regression are indicative of pcALCL (1). In this context, the size of the first tumour in our case suggests that it might have been a LyP. However, we could not determine whether the first tumour failed to regress spontaneously, as it was completely resected. The clinical presentation of our case suggests that differentiating between LyP and pcALCL might be difficult, and, thus, LyP and pcALCL are considered to represent different ends of a spectrum of primary cutaneous CD30+ LPD (1). In this respect, it might be better to refer to the first tumour as a CD30+LPD.
Finally, accumulating evidence has demonstrated that LyP is associated with lymphoma and haematological malignancies (1–5), suggesting that LyP might be considered to be a dermadrome (a skin manifestation of an internal disorder) that is particularly associated with haematological malignancies.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize