


1Institute of Genomics and Integrative Biology, Council for Scientific and Industrial Research, New Delhi, India, 2National Skin Centre, and 3Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, Singapore
Abnormally high production of melanin or melanogenesis in skin melanocytes results in hyperpigmentation disorders, such as melasma, senile lentigines or freckles. These hyperpigmentary skin disorders can significantly impact an individual’s appearance, and may cause emotional and psychological distress and reduced quality of life. A large number of melanogenesis inhibitors have been developed, but most have unwanted side-effects. Further research is needed to better understand the mechanisms of hyperpigmentary skin disorders and to develop potent and safe inhibitors of melanogenesis. This review summarizes the current understanding of melanogenesis regulatory pathways, the potential involvement of the immune system, various drugs in current use, and emerging treatment strategies to suppress melanogenesis.
Key words: skin pigmentation; melasma; melanin; tyrosinase.
Accepted Jul 3, 2018; Epub ahead of print Jul 4, 2018
Acta Derm Venereol 2018; XX: XX–XX.
Corr: Navin Kumar Verma, Lee Kong Chian School of Medicine, Dermatology and Skin Biology, Nanyang Technological University Singapore, 59 Nanyang Drive, Experimental Medicine Building, Singapore 636921. E-mail: nkverma@ntu.edu.sg; Hemant K. Gautam, Institute of Genomics and Integrative Biology, Council for Scientific and Industrial Research (CSIR-IGIB), Sukhdev Vihar, Mathura Road, New Delhi 110 025, India. E-mail: hemant@igib.res.in
Skin hyperpigentation disorders, including melasma, solar lentigines and freckles, are common dermatological concerns. These disorders are caused by excessive accumulation of melanin in the skin through a process called melanogenesis. While there are a diverse range of therapeutic modalities available to manage skin hyperpigmentation, there is an ongoing quest to develop more potent and safe inhibitors of melanogenesis. Here we provide an overview of the current understanding of melanogenesis pathways and review various inhibitors that are currently being applied or investigated to target melanogenesis.
Melanogenesis is a multistep physiological process that results in the synthesis of a complex darkly-pigmented biopolymer called “melanin”. Melanin is synthesized by melanosome, a lysosome-related organelle in melanocytes, and helps protect the skin from the harmful effects of sunlight, toxic drugs and chemicals (1, 2).
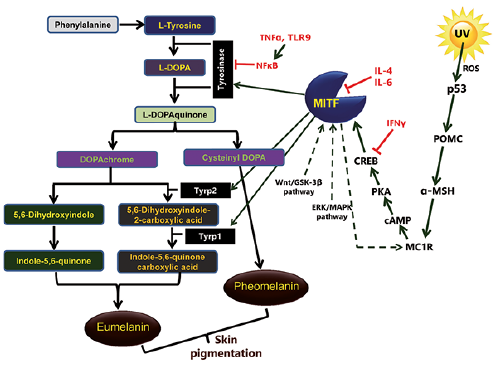
There are 2 types of melanin; eumelanin and pheomelanin. In the melanogenesis pathway, 3 main enzymes are involved; tyrosinase, tyrosinase-related protein 1 (Tyrp1, also known as gp75 glycoprotein or gp75) and tyrosinase-related protein 2 (Tyrp2, also known as dopachrome tautomerase or Dct). The melanogenesis process is initiated either by the hydroxylation of phenylalanine into L-tyrosine or directly by L-tyrosine, which is then hydroxylated to L-dihydroxyphenylalanine (L-DOPA). L-DOPA is further oxidized into L-DOPAquinone (DQ). Both of these reactions are catalysed by tyrosinase, which is therefore a key rate-limiting enzyme in melanogenesis.
The downstream pathway of melanogenesis involves intramolecular addition of an amino group to DQ, generating DOPAchrome. After the formation of DQ, the downstream melanogenesis pathway is divided into 2 parts, leading to the synthesis of “black-brownish eumelanin” and “red-yellow pheomelanin”. In the eumelanogenesis pathway, the DOPAchrome is either spontaneously converted to 5,6-dihydroxyindole or enzymatically converted to 5,6-dihydroxyindole-2-carboxylic acid by Tyrp2. Finally, polymerization of indole and quinones results in the formation of eumelanin. Pheomelanin synthesis is dependent on the presence of cysteine, which reacts with DQ to form cysteinyl-DOPA, and is further converted into quinoline, then, finally, polymerizes to pheomelanin (Fig. 1). While melanin plays a key role in protecting the skin from harmful ultraviolet (UV) radiation, abnormally high production and accumulation of melanin in the skin can lead to hyperpigmentation disorders.
Fig. 1. A schematic generalized signal transduction cascade of melanogenesis in humans.
While hyperpigmentation of the skin is usually harmless, increased pigmentation, especially on the face, such as melasma, solar lentigines and freckles, presents a significant cosmetic nuisance and can cause distress to the affected individual. Melasma presents with dark patches distributed symmetrically on the face and neck and hyperpigmented macules over the skin (Fig. 2A). Freckles are flat, small tan, predominantly present on the face, although other sun-exposed areas of the skin are at high risk (Fig. 2B). Lentigines are marked by the presence of a small brown patch, a benign lesion that mostly occurs on sun-exposed areas (Fig. 2C). Lentigines and freckles differ in terms of the number of melanocytes. In freckles, the number of melanocytes remains the same, but there is an increased amount of melanin; whereas, lentigines result from an increased number of melanocytes.
Fig. 2. Examples of human skin disorders caused by hyperpigmentation. (A) Melasma, (B) freckles, and (C) senile lentigines. A written permission is given to publish these photos.
Currently, hyperpigmentation disorders are treated with a wide range of topical hypopigmenting or skin-lightening agents, chemical peels, laser therapy, cryotherapy and superficial dermabrasion. Combination therapeutics is the preferred mode of treatment for the management of these conditions, which allows synergism and reduces the chances of untoward effects. For example, the most popular triple combination, containing hydroquinone, tretinoin and fluocinolone acetonide, is considered as one of the gold-standard treatments for melasma. This triple combination has also been found to enhance the resolution of solar lentigines and to significantly reduce melanin levels and lentigines count (3). However, these treatment modalities do not completely eradicate skin lesions. Thus, there is an increasing need to develop alternative, more specific and effective, treatment options.
The search for an effective inhibitor of melanogenesis has led to the discovery of hundreds of natural and synthetic compounds with potential anti-melanogenic activities (4). Better understanding of the melanogenesis regulatory pathways may help determine more specific target(s) for existing or new drugs, which could be used to regulate these pathways and control hyperpigmentation disorders.
Melanogenesis is regulated through a series of multistep signal transduction cascades associated with the enzymes tyrosinase, Tyrp1 and Tyrp2 (Fig. 1), as described below.
UV radiation-induced tumour suppressor protein p53 critically regulates the expression and activity of tyrosinase and Tyrp1 (5). The promoter of proopiomelanocortin (POMC) contains the p53 consensus sequence, where p53 binds and regulates the expression of this gene. UV exposure-induced activation of p53 results in increased expression of POMC, which is then cleaved into small peptides, such as ACTH, α-, β-, and γ-MSH. The POMC-derived α-MSH stimulates the melanocortin-1 receptor (MC1R) on melanocytes, resulting in increased production of eumelanin. In addition, UV radiation enhances the production of reactive oxygen species (ROS) in keratinocytes and melanocytes, and at high concentration ROS causes DNA damage, further activating p53, and thus triggering melanogenesis (6).
Microphthalmia-associated transcription factor (MITF) is a transcription factor, which contains a basic helix-loop-helix-leucine zipper (bHLH-LZ) structure and acts as a master regulator of melanocyte development, survival and its functions (7). It regulates the key melanogenic enzymes tyrosinase, Tyrp1 and Tyrp2. Multiple signalling pathways have been identified that are involved in MITF regulation as follows:
cAMP-dependent signalling pathway. Once the MC1R receptor on melanocytes is stimulated by α-MSH, it triggers production of cAMP via the activation of adenylyl cyclase. cAMP further activates the protein kinase A (PKA), which phosphorylates the cAMP response element binding protein (CREB), leading to the up-regulation of MITF. MITF regulates transcription of crucial pigmentary genes coding melanogenesis-related proteins (MRPs) through interaction with the M- and E- boxes present in promoter regions of tyrosinase, Tyrp1 and Tyrp2.
Wnt or β-catenin pathway. Wnts are cysteine-rich glycoproteins and play a crucial role in melanocyte development. It binds to the G-protein-coupled receptor “Frizzled” and inactivates the glycogen synthase kinase-3β (GSK-3β), causing the accumulation of β-catenin in the cytosol. β-catenin translocates into the nucleus, where it increases the expression of MITF.
The inhibition of mitogen-activated protein kinase (MAPK) kinase inhibits MITF turnover in the presence of cycloheximide (8), thus implicating extracellular signal-regulated kinase (ERK)/MAPK signalling in MITF degradation. In this pathway, ligand-receptor interactions activate the Ras protein, which further activates B-Raf kinase and consequently ERK1/2. MAPK phosphorylates MITF protein leading to the ubiquitination and degradation of MITF resulting in decreased expression of the melanogenesis-related genes.
NO/cGMP signalling pathway. The involvement of nitric oxide (NO)/cGMP pathway in UV-B-mediated melanogenesis was first discovered in the ink gland of Sepia officinalis. The excitatory neurotransmitter L-glutamate interacts with N-methyl-D-aspartate (NMDA) receptors and stimulates nitric oxide synthetase (NOS) to produce NO. NO further activates its downstream target guanylate cyclase to elevate cGMP, which activates the cGMP-dependent protein kinase (also called Protein Kinase G or PKG). PKG, as well as cGMP, phosphorylate and activate CREB, which further regulates the expression of MITF. MITF after activation up-regulates the expression of melanogenic genes, resulting in increased melanin production.
The cutaneous response to UV-B, mediated by the MC1R, affects the expression of the mitochondria-associated genes (9). In particular, MC1R-antagonist agouti down-regulates the expression of genes responsible for the mitochondrial redox ability in melanocytes (10). Moreover, the mitochondrial protein prohibitin is a cellular target of compounds that up-regulate pigmentation in wild-type and albino melanocytes (11). The mitochondrial dysfunction is associated with acquired or inherited pigmentary diseases, such as vitiligo and albinism (12). Mitochondria physically interacts with melanosome through fibrillar bridges which are modulated by Mfn2, a mitochondrial membrane protein involved in mitochondrial fusion, maintenance and the operation of the mitochondrial network (13). Furthermore, mitochondrial fission suppresses α-MSH-induced hyperpigmentation and mitochondrial fusion enhances melanin production. Melanogenesis suppression by mitochondrial fission occurs via activation of the ERK pathway (14), which results in phosphorylation and subsequent degradation of MITF (Fig. 3). However, the exact mechanism by which the mitochondrial fusion enhances melanogenesis is not clear.
Fig. 3. The mechanism by which mitochondrial dynamics regulates melanogenesis. Mitochondrial fission causes increased production of reactive oxygen species (ROS), resulting in activation of a ROS-ERK signalling pathway, which further induces the phosphorylation of microphthalmia-associated transcription factor (MITF). The phosphorylation of MITF causes its ubiquitination and proteasomal degradation, which subsequently downregulates melanogenesis. In contrast, mitochondrial fusion upregulates melanogenesis via an unknown mechanism. DRP1, FIS1, and MTP18 are fission proteins while OPA1, MFN1, and MFN2 are fusion proteins.
Increasing evidence has established that melanocytes are active factors in the skin immune system, play an essential role in immune responses, and have immunomodulatory properties (15, 16). On the other hand, immune mediators, such as cytokines, directly or indirectly regulate the proliferation, differentiation of melanocytes and melanogenesis (Fig. 1). For example, IL-4 inhibits melanogenesis and down-regulates the expression of melanogenesis-associated genes through activation of JAK2-STAT6 pathways (17). IL-6 suppresses melanogenesis by reducing the expression of tyrosinase and MITF transcript (18). TNF-α inhibits melanogenesis mainly via nuclear factor kappa B (NF-κB) and reduces the half-life of tyrosinase (19). Interferon-gamma (IFN-γ) has been implicated in the pathogenesis of vitiligo, where it is known to induce apoptosis in melanocytes (20). It inhibits melanogenesis by altering the expression of melanogenic enzymes and inhibiting the binding of CREB to MITF promoter via STAT1. In addition, IFN-γ maintains skin pigmentation homeostasis and mediates hypopigmentation via IRF1, which further controls melanosome maturation in melanocytes (20).
Toll-like receptors (TLRs), which recognize pathogen-associated molecular pattern (PAMP) present in microbes, negatively regulate the melanogenesis (21). Human melanocytes constitutively express mRNA and protein for TLR2, 3, 4, 5, 7, 9 and 10, and these TLRs play essential roles in the modulation of melanogenesis (21). However, the exact mechanism by which TLRs control melanogenesis is not clearly understood. A recent study demonstrates that TLR9 regulates melanogenesis via the activation of NF-κB (22).
Since tyrosinase is the key enzyme in the melanogenesis pathway, most earlier studies have focused on inhibiting the activity of this enzyme in order to control melanogenesis. Tyrosinase is a copper-containing, membrane-bound glycoprotein located in the melanosome membrane. It has an inner melanosomal domain that contains the catalytic region, a short transmembrane domain and a cytoplasmic domain. The majority of hypopigmenting agents act directly or indirectly on tyrosinase and inhibit its activity via competitive, uncompetitive or non-competitive mechanisms (23).
Tyrosinase inhibitors obtained from plants. Several molecules, such as phenolics, terpenes, steroids, chalcones, flavonoids, alkaloids, long-chain fatty acids and coumarins are derived from plants (23). These natural molecules are able to slow down the melanogenesis process by reversibly or irreversibly inhibiting the enzyme activity. There are a large number of plant-derived melanogenesis inhibitors, including hydroquinone, arbutin and aloesin, currently being used as skin-lightening agents (Table I). Hydroquinone has been widely used for more than 40 years as a skin-whitening agent; however, this compound causes skin irritation, induces mutations in mammalian cells, and is cytotoxic to mammalian cells (24). After hydroquinone, the second most commonly used pharmacological agent is retinoid. Retinoid was initially used in combination with hydroquinone to enhance its effect (25), but later it was discovered that retinoid has its own effect on pigmentation. In particular, retinoids, such as adapalene and tretinoin have been used successfully in lentigines (26, 27). Both tretinoin and vitamin A acid inhibit the activity of tyrosinase and interfere with pigment transfer to keratinocytes (28, 29). The other commonly used compound is arbutin, which is a derivative of hydroquinone and is found in blueberries, cranberries, wheat and pears. Arbutin causes reversible inhibition of tyrosinase activity (30) with fewer side-effects compared with hydroquinone. Aloesin (2-acetonyl-8-beta-D-glucopyranosyl-7-hydroxy-5-methylchromone) is another natural compound, obtained from aloe vera (Aloe barbadensis miller), which competitively inhibits tyrosinase (31). This compound is relatively safe (31) rendering it a promising active ingredient as skin whitening agent for cosmetic applications.
Table I. Melanogenesis inhibitors
Tyrosinase inhibitors obtained from microorganisms. Several microorganisms, including bacteria and fungi, are known to produce anti-melanogenic compounds. Currently, many such molecules have been discovered and tested for their anti-melanogenic activity. Kojic acid (5-hydroxy-2-hydroxy-methyl-4H-pyran-4-one) is a naturally occurring metabolite obtained from Acetobacter, Aspergillus and Penicillium (32). It is hydrophilic in nature and acts as a chelating agent for Cu2+ in the active site of tyrosinase. In addition, it suppresses the tautomerization of dopachrome to 5, 6-dihydroxyindole-2-carboxylic acid (32). Azelaic acid (1, 7-heptanedicarboxylic acid) is a reversible competitive inhibitor of tyrosinase, which is produced by Pityrosporum ovale. The azelaic acid in combination with an antioxidant compound, taurine, inhibits tyrosinase through activation of the ERK pathway (33).
Synthetic tyrosinase inhibitor. Besides the natural sources, tyrosinase inhibitors are also chemically synthesized or modified by many pharmacological industries. Chemical modifications, such as replacement of the functional groups, make these molecules more efficient and potent than their parent compound (23).
Resveratrol derivatives. Resveratrol is a polyphenolic compound that reduces the expression of tyrosinase, Tyrp1, Tyrp2, and MITF, and thus decreases hyperpigmentation. New synthetic analogues of resveratrol have been developed by replacing the CH group with a nitrogen atom, which has an increased tyrosinase inhibitory effect (34).
Cinnamic acid derivatives. Cinnamic acid is a moderate inhibitor of the tyrosinase enzyme (IC50 2.1 mM). Derivatives of cinnamic acid have shown an increased inhibitory effect against tyrosinase with lower IC50 values (35). This increased potency of cinnamic acid derivatives is due to the accelerated hydrophobic interactions between the hydrophobic side-chains of the ligands and the long lipophilic narrow gorge located near to the active site of the enzymes (35).
Polyphenol derivatives. The polyphenol derivatives represent a large and diverse group of a compound containing one or more phenolics. Alteration in the presence and position of additional substituents on the backbone structure results in a change in activity of these compounds. For example, benzylbonzoates, derived by changing the position of a hydroxyl group on 1 of the 2 aromatic rings, resulted in an increased inhibitory effect on the tyrosinase.
Kojic acid (KA) derivatives. KA inhibits tyrosinase activity by chelating metal ions, such as Cu2+, which are required for the functioning of the enzyme. The biological activity of KA is due to its γ-pyranone structure. KA derivatives, developed by adding peptide and C-terminal amide group, have shown superior storage stability compared with KA (36). This increased activity of KA derivative is due to hydrophobic interactions between the side-chain of KA-AA-NH2 and tyrosinase active site (36).
Methanol derivatives. More recently, a group of researchers has designed and synthesized 17 (2-substituted phenyl-1,3-dithiolan-4-yl) methanol derivatives (37). These compounds have been found to inhibit the tyrosinase activity even more than KA or arbutin. Methanol derivatives bind to the active site of the tyrosinase and competitively inhibit its activity (37). In addition, tranexamic acid has been found to suppress melanogenesis and inhibit hyperpigmentation via the inhibition of tyrosinase; however, the efficacy of such a modality is yet to be established (38).
Post-translational modification of MITF protein reduces its expression. Post-translational modification of MITF, such as sumoylation, ubiquitination, and phosphorylation, play a major role in the regulation of the stability and the function of this protein. Sumoylation of MITF at specific amino acids, such as K182 and K316, has been shown to reduce the synergistic transcriptional activity of MITF at the Tyrp2 and melastatin/TRPM promoter (39, 40).
Ubiquitination has several effects on proteins’ activity and stability. It can cause the degradation of the protein via the proteasomal pathway, alter the cellular location of the protein, change their activity, and promote/prevent protein interactions. Ubiquitination of MITF at K201 causes its degradation via proteasome complex and mutation of this amino acid residue or inhibition of the proteasomal complex results in stabilization and accumulation of the MITF protein (7). In addition, phosphorylation of MITF at Ser73 via MAPK signalling results in the degradation of MITF and phosphorylation at Ser409 via c-kit signalling causes short-lived activation and then the destruction of MITF (41). However, phosphorylation at Ser298 by GSK-3β enhances the function of MITF (42).
MITF modulator acting through the signalling pathway. The expression of MITF is regulated through multiple signalling pathways and there are a large number of inhibitors that modulate these pathways. For example, AP736, a novel adamantyl benzylbenzamide derivative, inhibits the MITF transcripts by blocking the cAMP-PKA-CREB pathway (43, 44). The p21-activated kinase 4 (PAK4) regulates CREB and an inhibition of PAK4 downregulates the CREB protein, which sequentially decreases MITF and tyrosinase levels in melanocytes (45). Likewise, many inhibitors act on Wnt/β-catenin pathway to suppress melanogenesis. For example, the pyridinyl-imidazole compound causes a significant reduction in the transcription factor lymphoid enhancer-binding factor 1 (Lef1). Lef1, along with T-cell factor (Tcf), regulates gene expression by Wnt signalling. Imidazole treatment causes the reduction in Tcf/Lef levels and targets genes that are involved in Wnt signalling, such as Axin2, Lef1, MITF and Tyrp2 (46). Cardamonin, a chalcone from Alpinia katsumadai, inhibits melanogenesis by inducing GSK-3β-independent degradation of intracellular β-catenin (47). The acetylsalicylic acid (ASA) phosphorylates MITF via ERK pathway and thus triggers proteasomal degradation through enhanced ubiquitination (48). Shogaol, an active ingredient of ginger, acts in the same way, by activating the ERK that leads to the suppression of MITF expression (49). Gallic acid treatment of melanocytes activates the ERK/MAPK and PI3K/Akt pathways and inhibits the Wnt/β-catenin pathway (50). A role of COX-2 in the regulation of MITF and tyrosinase has also been reported (51).
The interfering RNA (RNAi)-mediated inhibition of MITF reduces melanogenesis. RNA interference mechanism is one of the best strategies to silence the expression of any gene of interest and is widely being explored for therapeutic applications. For example, transfection of melanoma cells with siRNA against MITF resulted in significant reduction in tyrosinase, Tyrp1 and MC1R levels, and melanin content was found to be significantly reduced up to ~50% compared with controls (52). However, there are challenges associated with siRNA therapeutics, including the stability and efficient delivery of siRNA molecules inside melanocytes, which limit the clinical translation of such an effective approach. Regulation of MITF expression through microRNAs, including miR-137, miR-148 and miR-182, has also been reported in many studies (53–56). A recent study suggests the potential of exploiting cell-permeable GapmeR RNAi technique (57) to specifically target melanogenesis pathway in melanocytes.
Melanocytes express an H+/K+ ATPase at the cytoplasmic membrane and melanosome contain a vacuolar-type H+ ATPase. Proton pump inhibitors (PPIs) inhibit gastric acid secretion via blocking the gastric H+/K+ ATPase, and this action triggers or worsens vitiligo by increased destruction of melanocytes by inducing apoptosis in patients with vitiligo (58). Omeprazole, a PPI used to treat gastroesophageal reflux disease, has been shown to inhibit melanogenesis by blocking ATP7A, which is a copper-transporting P-type ATPase that delivers copper to tyrosinase within the secretory pathway (59). Other PPIs, such as rabeprazole, lansoprazole, pantoprazole and esomeprazole, have also been found to inhibit melanogenesis via their copper-chelating activities without inducing cytotoxicity (60).
Sugars are the ubiquitous natural compound in humans and other organisms. The role of sugar and sugar-related agents in reducing melanogenesis process has been known for a quite long time (61, 62). These agents act on melanogenesis process via two different mechanisms. First, natural sugars create high osmotic potential, which affects the biogenesis of intracellular cargo and lysosomal and endocytic compartments. The proper maturation of melanosome is dependent on organelle formation and transport of melanogenesis-related proteins. Therefore, osmotic stress induced by the natural sugars influences the process of melanogenesis and the consequences results in hypopigmentation.
Secondly, sugar-derivatives inhibit tyrosinase maturation. The tyrosinase enzyme requires N-glycosylation at 7 different sites for its proper functioning and localization, which is a multistep process that occurs mainly in 2 cellular compartments: (i) endoplasmic reticulum, where N-acetyl glucosaminyl attaches carbohydrate to tyrosinase and (ii) Golgi apparatus, where α-glucosidases and α-mannosidases detach the carbohydrate groups. If the activity of these enzymes is impaired, tyrosinase undergoes proteolytic degradation, which results in hypopigmentation. Several attempts are being made to use glycosylation inhibitors to regulate the process of melanogenesis (62). In particular, two specific inhibitors, deoxynojirimycin (α-glucosidase inhibitor) and deoxymannojirimycin (α-mannosidase inhibitor), were found to be effective in decreasing melanin production (62). However, these glycosylation inhibitors have unwanted cytotoxic effects.
The regulation of melanogenesis is highly complex and a number of cellular factors are involved in this process. Over the past few years, there have been remarkable advances in the understanding of melanocyte biology. This area of research has led to the discovery of a diverse and complex regulatory mechanism of melanogenesis. A large number of melanogenesis inhibitors has been developed, and their mechanisms have been identified, but the major concern with these inhibitors is their lack of target specificity, toxicity, and high frequencies of adverse reactions. Better understanding of the regulatory mechanism of melanogenesis will help researchers to develop a novel target to modulate this process. Ongoing research in the area of structural characterization of melanogenesis pathway mediators, in silico analysis and emerging nanotechnological approaches coupled with gene silencing techniques would provide molecularly targeted inhibitors for next-generation therapeutics in skin hyperpigmentation.
This work was supported in part by grants from Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore Start-Up Grant to NKV and CSIR, TOUCH grant, BSC0302 to HKG.


 Click to show fullsize
Click to show fullsize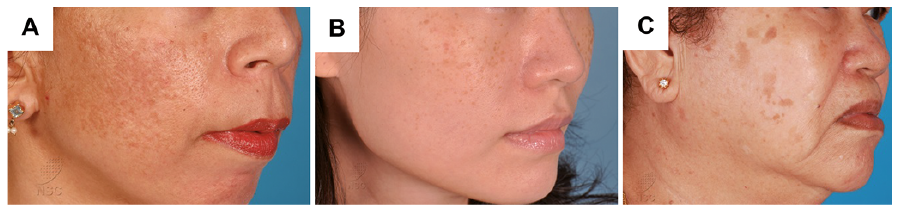 Click to show fullsize
Click to show fullsize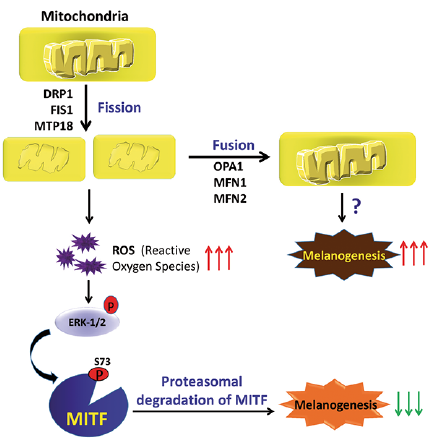 Click to show fullsize
Click to show fullsize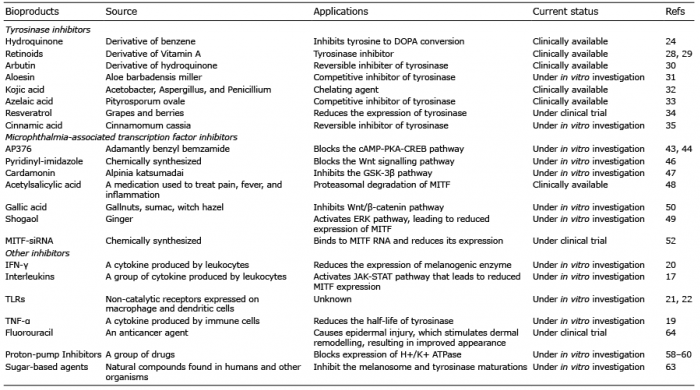 Click to show fullsize
Click to show fullsize