


1Laboratory of Molecular and Cell Biology, 21st Dermatology Division, Istituto Dermopatico dell’Immacolata, IDI-IRCCS, and 3Genetic and Rare Diseases Research Area, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy. E-mail: d.castiglia@idi.it
Accepted Aug 7, 2018; Epub ahead of print Aug 7, 2018
Darier disease (DD) is a rare skin condition with autosomal dominant inheritance that manifests in childhood or adolescence (1, 2). It is characterized by keratotic papules that may coalesce into large warty plaques, predominantly affecting seborrhoeic areas, such as the forehead, central chest, back and scalp borders, usually accompanied by palmoplantar pits and characteristic nail changes (i.e. longitudinal leukonychia and V-shaped notches at the distal end of the nail plate) (1, 3). Involvement of intertriginous areas and psychiatric symptoms may also be present (1, 3). Heat or humidity, excessive sweating, exposure to ultraviolet (UV) light, friction, medications and infections, as well as pregnancy or delivery, aggravate the disorder (3). Histologically, DD is characterized by loss of adhesion between epidermal cells, termed acantholysis, leading to suprabasal cleavage and abnormal keratinization, which manifests as corps ronds and grains (4).
DD is caused by mutations in the ATP2A2 gene, which encodes isoforms of the sarco/endoplasmic reticulum ATPase type 2 (SERCA2) in keratinocytes and cells from other tissues (5). SERCA2 is a calcium pump incorporated in the endoplasmic reticulum (ER) membrane that regulates Ca2+ transport from the cytoplasm into the ER lumen. It is organized in 10 transmembrane helices (M1–M10) intercalated by 5 stalk sectors (S1–S5), and in a cytosol-exposed region comprising the phosphorylation and ATP-binding domains (5). In 2000, genetic mosaicism for mutations in ATP2A2 was demonstrated to cause the segmental form of the disease (6), which is quite rare. Segmental DD can manifest in a mosaic pattern distinctive of 2 disease subtypes: segmental DD type 1, which is characterized by warty papules distributed in a linear pattern on a background of clinically normal skin, and segmental DD type 2, in which linear DD lesions are usually more severe and may manifest early after birth, superimposed on a background of generalized DD (7, 8). Segmental DD type 1 was hypothesized to result from a de novo postzygotic ATP2A2 mutation that occurs during embryogenesis, and a few studies have provided genetic proof for this aetiology (4, 9), whereas a loss-of heterozygosity (LOH) in a heterozygous individual was hypothesized for segmental DD type 2 (10). Although there is no direct evidence for LOH as a causal event for DD type 2, molecular genetic support has been provided (8). We report here on the molecular basis of the mosaicism in an Italian woman with segmental DD type 1.
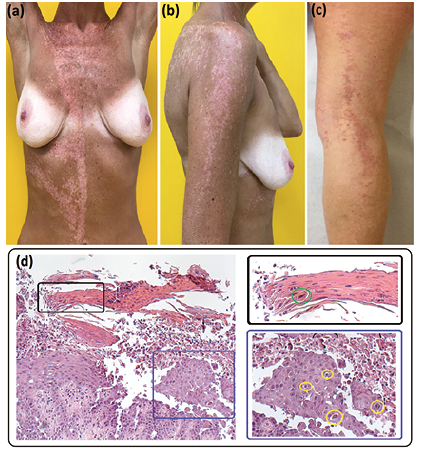
A 47-year-old woman presented with keratotic papules on the right side of her trunk, and right arm and leg. The lesions appeared on her chest when she was 27-year-old and spread to the right arm and leg 8 years later, during her first pregnancy. She reported worsening symptoms during summer and spontaneous improvement in winter. None of her family members had similar cutaneous manifestations. Examination showed a linear grouping of papules following Blaschko’s lines on the right arm, thigh and leg, and on the right side of the dorsum, abdomen and chest, where lesions also extended beyond the midline (Fig. 1a–c). No oral, nail or hair abnormalities were found. A skin biopsy taken from a lesion of the posterior aspect of the right leg showed suprabasal acantholytic dyskeratosis with corps ronds and grains (Fig. 1d). Based on the clinical presentation and histopathological findings a segmental DD type 1 was suspected. Treatment included topical steroids, antibiotics and emollients, followed by tazarotene 0.05% and, finally, isotretinoin 0.05%, with only temporary symptom relief.
Fig. 1. Clinical and histopathological findings. (a, b) Hyperkeratotic papules following the Blaschko’s lines over the right side of the patient’s trunk and her right arm. (a) Note the sharp midline demarcation on the abdomen, clearly visible after sun exposure during the summer, on a background of post-inflammatory hypopigmentation. (c) Hyperkeratotic papules on the right leg during the winter. (d) Hyperkeratosis with parakeratosis, acanthosis and suprabasal acantholysis in the haematoxylin-eosin staining. Higher magnification shows dyskeratotic cells with a pyknotic nucleus surrounded by a halo, termed corps ronds, within the spinous layer (d: blue inset, yellow circles), and flattened dyskeratotic cells with an elongated nucleus, termed grains, within the parakeratotic horny layer (d: black inset, green circle). Haematoxylin-eosin staining (d) × 200 (left panel).
To confirm the clinical diagnosis, a search for the post-zygotic ATP2A2 causative mutation was undertaken, following written informed consent. Genomic DNA samples purified from both patient blood and separated epidermis obtained by dispase II-treated biopsies from affected (right thigh) and non-involved skin (left buttock) were subjected to a TruSight sequencing panel (Illumina), which was then directly interrogated for mutations in the ATP2A2 gene. This analysis identified the deletion c.2256_2258delCTA, p.Tyr753del (NM_170665.3, NP_733765.1) in 14 sequence reads-out of 44 across the DNA extracted from the affected epidermis (Fig. S1a), and excluded this change in the DNA from non-involved epidermis and blood (read depth: 55). Subsequent Sanger sequencing performed on different DNA aliquots confirmed the presence of the heterozygous c.2256_2258delCTA in the DNA of the affected epidermis only (Fig. S1b). The mutation was not found in the blood DNA of the patient’s 12-year-old son.
We report here the 7th case of molecularly characterized segmental DD (4, 6, 8, 9, 11) (Table SI). To our knowledge, the p.Tyr753del somatic mutation is not reported in the literature and not annotated in any human genetic variant databases. The PROVEAN predictor for mutation pathogenicity (http://provean.jcvi.org/seq_submit.php) classifies it as deleterious (score: –15.716; cut-off: –2.5). The p.Tyr753del affects both SERCA2 isoforms at the transition of the stalk domain S5 to the transmembrane M5 helix. It is similar in its consequences to the p.Ile752_Tyr753delinsAsn and to other missense and single amino acid deletion mutations that fall around residue 753 causing non-segmental DD (2, 12). The massive parallel sequencing approach used here to molecularly characterize a mosaic DD reveals that the mutant allele in the affected epidermis accounts for approximately 31.8% (14/44) of the sequence reads (i.e. well below the 50% rate expected for all cells being heterozygous for the mutation). It has been demonstrated previously that a threshold number of mutated cells is required for the development of clinical manifestations (4). Our findings show that a percentage of mutant cells close to 30% is sufficient to induce cutaneous DD lesions. Nucleotide peaks in the Sanger sequencing traces confirmed that the p.Tyr753del mutant alleles are under-represented compared with the wild-type allele. Moreover, Illumina sequence reads did not detect the mutation in the patient’s blood, indicating that it had arisen after differentiation of the embryonic epiblast when ectoderm and mesoderm diverge in distinct layers (4).
In conclusion, this report demonstrates that the spontaneous somatic p.Tyr753del mutation is the cause of the segmental phenotype in our patient. She was informed about the possibility that the mosaic condition could also involve the gonads, with a risk of transmitting the full-blown disease to her children. Absence of the mutation in the blood DNA of the patient’s son allowed us to exclude clinical onset of DD in this individual.
Authors acknowledge the support from the Italian Ministry of Health (Ricerca Corrente 2018-2020).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize