


Department of Dermatology, Ehime University Graduate School of Medicine, 791-0295 Toon, Ehime, Japan. E-mail: kshira@m.ehime-u.ac.jp
Accepted Aug 31, 2018; Epub ahead of print Sep 3, 2018
Acquired partial lipodystrophy (APL) is a rare disorder characterized by the loss of subcutaneous adipose tissue, usually starting in the face and extending to the neck, arms, and trunk (1). The exact pathogenesis remains unclear, but it can be associated with metabolic disorders and autoimmune diseases, particularly juvenile dermatomyositis (JDM) (2). Here we report a rare case of APL in an adult patient with anti-Mi-2 antibody positive dermatomyositis who did not develop metabolic abnormalities.
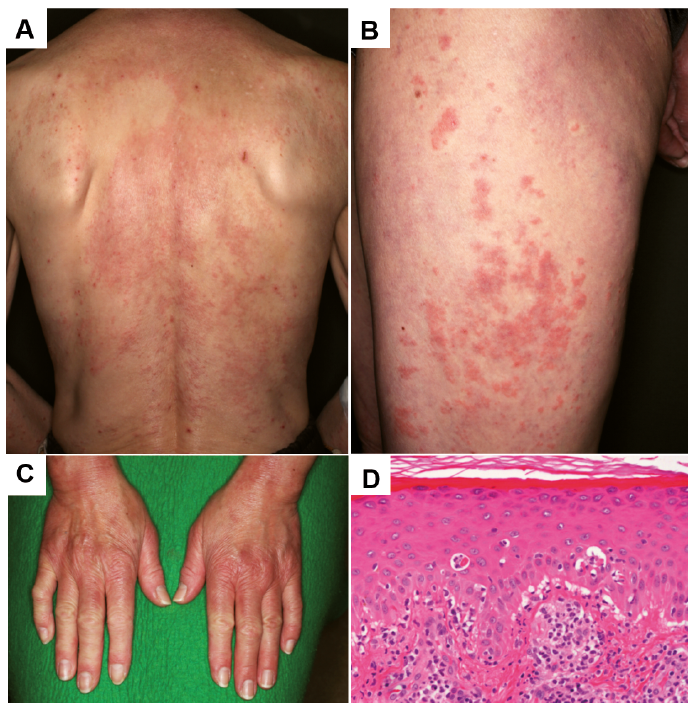
A 53-year-old woman presented to our institution with a 3-month history of erythematous eruptions on the face, upper trunk, extremities, and dorsal surfaces of both hands. Physical examination revealed diffuse edematous erythema on the back and thighs (Fig. 1A, B). We also detected erythematous lesions on the dorsal surfaces of both hands, periorbital erythema and Gottron papules (Fig. 1C), but no periungual telangiectasia was evident. The patient showed no evidence of myositis. Blood testing showed that complete blood count, liver and renal function test and urinalysis were all normal. The results of antinuclear antibody testing were positive, with a titer of 1:2560. C3 and C4 fractions of the complement were within normal limits. C3 nephritic factor (C3NeF) was not available in our hospital. Anti-ARS, anti-MDA5, anti-TIF-1γ, anti-RNP, anti-Sm, anti-ssDNA, and anti-dsDNA antibody tests all yielded negative results. Positive results were obtained for anti-Mi-2 antibody (> 150). No abnormalities in serum lipid or glucose levels were detected. Computed tomography demonstrated mild interstitial pneumonia.
Fig. 1. Diffuse edematous erythema on the back (A) and thighs (B). C) Erythematous lesions on the dorsal surfaces of both hands. Periorbital erythema and Gottron papules were also seen. D) Histopathological findings of biopsy tissues show interface dermatitis with vacuolar change with sparse superficial perivascular infiltration of lymphocytes. Hematoxylin-eosin stain, original magnification: ×100.
Skin histopathology from erythematous lesions showed interface dermatitis with perivascular infiltration of lymphocytes in the dermis (Fig. 1D), consistent with dermatomyositis. Cancer screens revealed no indication of malignancy and ruled out underlying neo-plastic disease. Since the patient showed no evidence of systemic disorder, organ damage and myositis, we followed-up skin lesions by topical steroid ointment. Skin lesions then started to gradually improve. However, after a 1-year follow-up period, she mentioned a progressive loss of fat in the facial region over the last several months, after her family pointed out changes in her face. She had no history of dieting, and had never used drugs associated with lipodystrophy. No family members had any history of connective tissue disease or lipodystrophy. Physical examination revealed a marked loss of adipose tissue in the face, particularly as bilateral loss of the buccal fat pads and prominence of the zygomatic arches and forehead (Fig. 2D–F). Loss of some fat on both proximal arms were also seen. The trunk and lower limbs appeared unaffected. Based on the above findings, the diagnosis of APL was made.
Fig. 2. A–C) Erythematous lesions on the face at first visit. D–F) The face of the patient after onset of lipodystrophy. A marked loss of adipose tissue is evident in the face, particularly as bilateral loss of buccal fat pads and prominence of the zygomatic arches and forehead.
Lipodystrophy is a group of adipose tissue disorders classified into genetic/acquired or generalized/partial lack of adipose tissue (lipoatrophy) (1). APL presents with late-onset symmetrical loss of fat from the face, neck, arms, and trunk. Females are affected four-times more often than males. In our case, the female patient showed severe loss of adipose tissue in the face and upper extremities with sparing of the lower extremities, consistent with APL.
APL has been associated with infections, antiretroviral therapy for the treatment of human immunodeficiency virus infection and autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, Sjögren syndrome, and dermatomyositis (3). Dermatomyositis is the autoimmune disease most frequently related to the development of lipodystrophy (4), but most cases are specifically associated with JDM. JDM is characterized by constitutional symptoms, pathological calcifications, skin infarctions, lipodystrophy, and a lack of internal malignancy (5). The prevalence rates of lipodystrophy in JDM vary from 10% to 40% (2), but only 4 cases of lipodystrophy affecting adult-onset dermatomyositis patients have been reported to date (6–9). To the best of our knowledge, APL associated with adult-onset dermatomyositis with anti-Mi-2 antibodies has never been reported.
The etiology and pathogenesis of lipodystrophy associated with autoimmune disease remain unclear. No evidence suggests that standard therapies for autoimmune disease, such as systemic corticosteroids, cause lipodystrophy. According to previous reports, patients often have the polyclonal immunoglobulin C3NeF and show low C3 levels. C3NeF prevents C3bBb (C3 convertase) from being inactivated, leading to excessive consumption of C3 and activation of the complement alternative pathway. This mechanism has been postulated to be involved in the adipocyte lysis and the development of the disease (3). However, in our case, immunologic abnormalities such as low levels of C3 were not demonstrated. Metabolic abnormalities such as diabetes and hypertriglyceridemia are also well-recognized consequences of lipodystrophy (2). In dermatomyositis, these metabolic effects are theorized to be related to disrupted muscle modulation of insulin (10). However, metabolic abnormalities were also absent in our patient. We could not demonstrate the precise mechanisms of fat atrophy associated with dermatomyositis, but the sites of lipodystrophy were mostly the same or close to the areas showing severe skin lesions on her face (Fig. 2). Lipodystrophy may thus occur as a result of inflammation, as exemplified by a loss of subcutaneous tissue in lupus panniculitis. In most cases, patients developed lipodystrophy within a year after the onset of dermatomyositis as a late complication of severe, chronic disease, but one report presented a rare case in which lipodystrophy developed simultaneously with dermatomyositis (6). In our case, lipodystrophy arose one year after onset of dermatomyositis.
The progression of lipodystrophy is difficult to inhibit. According to some reports, oral prednisone and myco-phenolate mofetil do not improve lipodystrophy (8, 9). Walker et al. (11) demonstrated good cosmetic outcomes following rosiglitazone treatment in a young woman with APL. Thiazolidinediones are agonists of the peroxisome proliferator-activated receptor, which plays an important role in adipocyte differentiation. Future plastic surgery including a series of reconstructions should also be offered when necessary.
Here, we have presented a rare case of partial lipodystrophy associated with anti-Mi-2-positive adult-onset dermatomyositis. Follow-up is crucial for patients with dermatomyositis regarding loss of subcutaneous fat is important.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize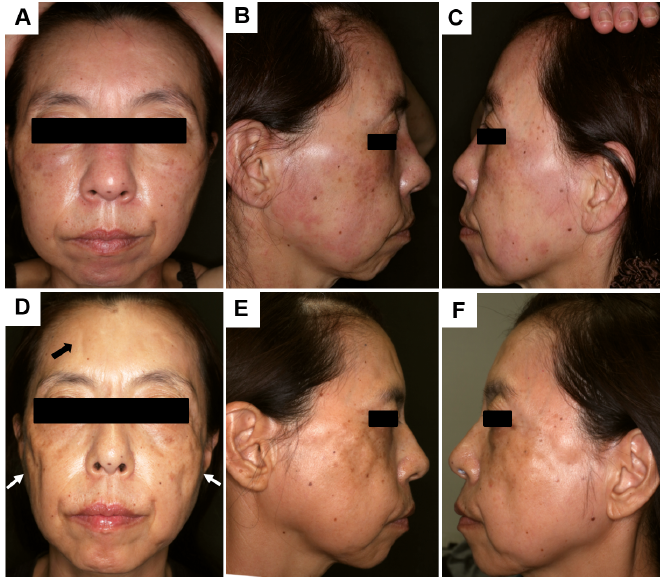 Click to show fullsize
Click to show fullsize