


Departments of 1Dermatology and Venereology and 3Pathology, The Second Hospital of Dalian Medical University, Dalian 116027, and 2Department of Dermatology and Venereology, Liaocheng People’s Hospital, Liaocheng, China. E-mail: wangaxdl@163.com
Accepted Sep 4, 2018; Epub ahead of print Sep 5, 2018
Langerhans cell histiocytosis (LCH) represents a group of rare and clinically heterogeneous conditions of unknown etiology (1). Although most LCH cases appear in pediatric patients, the disease does occur de novo in adults, with an estimated incidence of 1–2 per million. Most adult LCH cases involve only a single organ system, such as the bones or lungs. Diagnosis of LCH is generally dependent on biopsy and is made based on the observation of inflammatory cell infiltration with large numbers of histiocytic cells and Langerhans cells staining positive for S100 and CD1a (1). Here we report an unusual case of LCH involving the lymph nodes that presented with the clinical features of erythroderma.
A 75-year-old Chinese man had a 2-month history of pruritic papular eruption on his trunk and limbs that began following an exposure to rain. The rash had rapidly progressed to generalized exfoliative dermatitis in a 2-week period. The patient had experienced slight weight loss and fatigue for the previous one week but denied other systemic complaints including fever, expectoration, diarrhea, bone pain, and jaundice. Physical examination revealed enlarged bilateral axillary and inguinal lymph nodes exceeding 1 cm in diameter. Skin examinations revealed generalized uniform erythematous macules and diffuse scaling on the whole body (Fig. 1A, Fig. S1A). Blood tests revealed leukocytosis (14.61×109 cells/l) with a predominance of eosinophils (21.71%), anemia with low hemoglobin (119 g/l), and increases in the erythrocyte sedimentation rate, C-reactive protein and IgE (556 IU/ml) above the normal ranges. Chest X-rays and computed tomography (CT) scans showed mild interstitial changes. No Sézary cells were found in the peripheral blood. Routine studies included bacterial and fungal cultures from the skin and measurement of serum electrolyte, creatinine, and transaminase levels. Skull and abdominal CT scans showed normal results, and no bone alterations were observed.
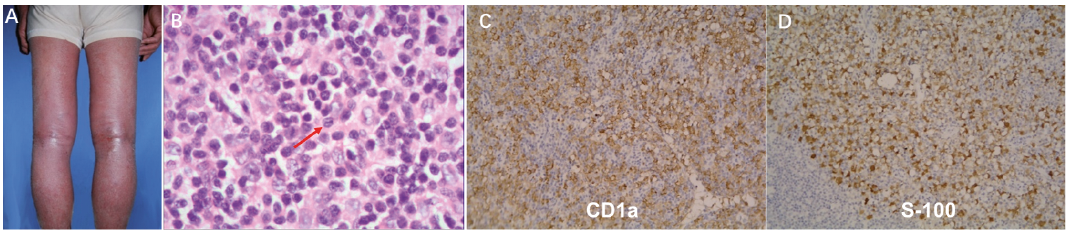
A skin biopsy of the affected area indicated hyperkeratosis, parakeratosis, acanthosis, and intercellular edema, with capillary dilation in the upper dermis and inflammatory infiltrate consisting of lymphocytes and eosinophils, collectively suggesting a case of superficial dermatitis (Fig. S1B, C). Histological examination of the inguinal node revealed destruction of the lymph node structure as well as tumor cell infiltration. Large cells with grooved nuclei containing compact chromatin along with eosinophils and plasma cells also were observed (Fig. 1B, Fig. S1D). These cells stained positively for S-100 protein and CD1a upon immunochemical analysis, and thus, a definitive diagnosis of LCH could be made (Fig. 1C, D).
Upon diagnosis of LCH, the patient was initially treated with oral methylprednisolone (40 mg/day) and application of a topical emollient to the affected areas daily. The sizes of the lymph nodes in the axillary and inguinal regions were significantly reduced after 2 weeks of these initial treatments, and the skin eruptions had improved. Improvements in the laboratory results also were noted. After 2 weeks of treatment, the methylprednisolone dose was tapered by 5 mg every 2 weeks to a maintenance dose of 5 mg/day. LCH in this patient responded completely to treatment, and thus, methylprednisolone use was discontinued after 3 months. No recurrence was observed during the 2-year follow-up.
Fig. 1. Langerhans cell histocytosis. (A) Generalized uniform erythematous macules and diffuse scaling affecting the patient. (B) Higher magnification images of lymph node showing large cells with grooved nuclei (arrows) and eosinophils. (H&E ×400). (C, D) Immunohistochemical staining revealed that the tumor cells were positive for CD1a (×200) and S-100 protein (×200).
Erythroderma, also known as exfoliative dermatitis, is an inflammatory reaction of the skin that is often secondary to a variety of causes (2). The most common causative factors are previous dermatoses, such as psoriasis and atopic dermatitis, followed by drug reaction, malignancy, or idiopathic causes. As this condition has received more attention, more unusual underlying dermatoses for erythro-derma have been reported. To our knowledge, our case presented is the third documented adult case of LCH involving the lymph nodes first manifesting as erythroderma (2, 3).
In general, drug therapy for erythroderma consists of topical and/or systemic medications based on the underlying etiologies (2). The severe generalized skin inflammation of erythroderma caused by LCH in adults is a rare entity, and due to the unclear causes and enigmatic pathogenesis of this rare entity, treatment strategies are based largely on empiric regimens.
The most appropriate treatment strategy for LCH remains controversial and varies according to the extent and severity of the disease at diagnosis. In 1990, the LCH Study Group divided LCH into two major categories: single-system LCH and multisystem LCH. Single-system LCH was further subdivided into single site (unifocal bone, skin or lymph node) and multiple site (multifocal bone or multiple lymph nodes) forms. Cases of single-system LCH, generally, have a high probability of spontaneous remission and a favorable outcome (4).
Considering the underlying immune mechanisms in the pathogenesis of LCH, corticosteroids should be the first choice. As expected, satisfactory remission was achieved with oral methylprednisolone alone in the presented case.
It is unclear how localized granulomatous inflammation of a lymph node progresses to an acute generalized inflammatory process in erythroderma. As our biological understanding of LCH and erythroderma has evolved, we have gained some insight into the pathogenesis of immune dysregulation in both diseases, including the role of the “cytokine storm” and elevated expression of adhesion molecules within lesions (4, 5). These findings suggest the possible application of a new class of drugs in the clinical treatment of LCH-related erythroderma. For example, tumor necrosis factor (TNF)-α is one of the most important inflammatory cytokines, and a TNF-α inhibitor, etanercept, was successfully used as salvage therapy in a child with multisystem LCH (6). Also, this type of drug has been used in the treatment of patients with erythrodermatic psoriasis (7). Alternatively, intravenous immunoglobulin (IVIG) combined with steroid therapy has been recently demonstrated to be an effective treatment for neurodegeneration observed in a case of LCH involving the central nervous system (8). IVIG has a variety of immune-modulating effects, and its protective actions include direct anti-inflammatory effects, scavenging of inflammatory mediators (e.g., cytokines), inhibition of complement formation, and modification of B-cell and T-cell functions (9, 10). Therefore, it can be postulated that a treatment regimen combining a TNF-α inhibitor and IVIG may be a promising alternative approach for LCH-induced erythroderma if the response to glucocorticoid therapy alone is insufficient.
Still additional drugs such as cladribine (2-CDA) and interferon-α have been shown to be effective for LCH treatment via their antiproliferative and immunomodulatory effects, although a well-organized prospective study is needed to confirm their efficacy (4).
Although single-system LCH in older patients usually follows a benign course (11), such patients should receive careful follow-up, given that LCH can progress to erythroderma, which can be life-threatening even when properly managed. For cases that do progress to erythroderma, once drug-related factors and underlying dermatoses are excluded, malignancy should be considered. In addition, both adult patients with LCH and erythroderma have an increased risk of a secondary malignancy. Although a tumor diagnosis was excluded during the period of hospitalization for the present case, the patient should continue to be monitored for a long time.
In summary, because of the rare morbidity and unspecific features of erythroderma secondary to LCH, its diagnosis in adults may be neglected during differential diagnosis. Histopathological biopsy should be performed to establish a diagnosis, especially in cases in which erythroderma secondary to LCH is clinically suspected. Unfortunately, the specific mechanisms of erythroderma and LCH have not been fully elucidated, and the mechanisms underlying the relationship between a localized granulomatous inflammation of lymph node tissue and an acute generalized inflammatory process in erythroderma need to be further investigated. For these reasons, early recognition of LCH manifesting as erythroderma is important for establishing proper treatment, and close surveillance for a prolonged period is necessary.
This work was supported by the National Natural Science Foundation of China (No. 81301365) and the Natural Fund of the Liaoning Provincial Science and Technology Department (No. 2014023011).


 Click to show fullsize
Click to show fullsize