


1Genetics and Rare Diseases Research Division, 2Pathology Division, Bambino Gesù Children’s Hospital-IRCCS, Piazza S. Onofrio 4, IT-00165 Rome, 3Laboratory of Clinical Pathology and Advanced Molecular Diagnostics, Istituto Dermopatico dell’Immacolata, IDI-IRCCS, and Università Cattolica del Sacro Cuore, Fondazione Policlinico Universitario Agostino Gemelli, 4Laboratory of Molecular and Cell Biology, Istituto Dermopatico dell’Immacolata, IDI-IRCCS, Rome, 5Pediatric Highly Intensive Care Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy, and 6Department of Dermatology, University Medical Center, Freiburg, Germany. E-mail: may.elhachem@opbg.net
#These authors contributed equally.
Accepted Sep 18, 2018; Epub ahead of print Sep 18, 2018
Epidermolysis bullosa simplex (EBS) is a phenotypically and genetically heterogeneous type of EB characterized by skin fragility and cleavage within the epidermis (1). The most common subtypes of EBS are due to dominant mutations in the KRT5 (keratin 5) and KRT14 (keratin 14) genes, but genetic defects in 10 additional genes are responsible for different variants. KLHL24 causative mutations have been identified in a novel EBS form characterized by denuded skin areas at birth and improvement in skin fragility with age (2, 3). KLHL24 encodes a member of the kelch superfamily, which includes proteins with variable tissue expression patterns involved in ubiquitination and proteasomal degradation of different substrates, including epidermal keratins (3, 4). It is notable that all of the 29 KLHL24 mutation-positive patients reported to date carried a heterozygous mutation in the first codon affecting translation initiation. The causal mutation shows a dominant pattern of inheritance in affected pedigrees or occurs as a de novo event (2, 3, 5–7). At present, the spectrum of clinical features and natural history of this EBS subtype remains incompletely characterized. We report here on 3 additional children with de novo KLHL24 codon 1 mutations, providing evidence for a wider clinical spectrum associated with these mutations.
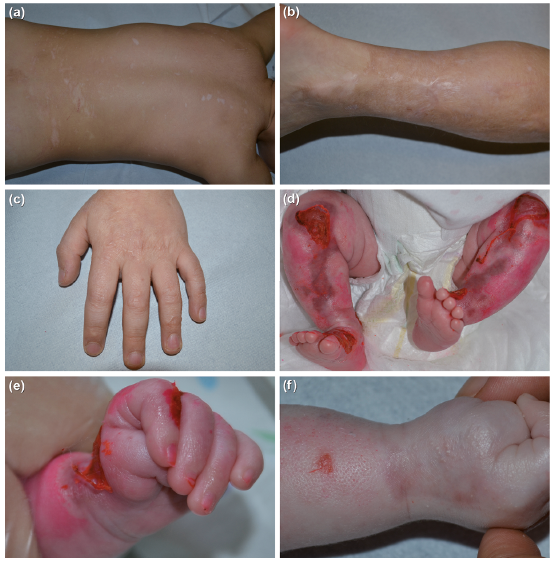
Case 1. Male, born from healthy parents at 37 weeks of gestation weighing 2,650 g. At birth, he presented extensive areas of denuded skin involving the limbs, buttocks, left mammary region, and oral lesions. The course was complicated by Serratia marcescens sepsis. Both immunofluorescence antigen mapping (IFM) and electron microscopy (EM) of a skin biopsy performed in a reference centre for EB showed normal expression of epithelial adhesion proteins in the absence of skin cleavage, and were thus considered uninformative. Skin erosions healed within the first month, leaving hypochromic, atrophic and raised linear-stellate scarring (Fig. 1a–c), and the patient did not show new blisters. Congenital erosive and vesicular dermatosis healing with reticulated supple scarring was diagnosed. In the following years, the patient developed occasional trauma-induced erosions, follicular atrophy, and dystrophy of toenails, which appeared thinned, brittle with longitudinal ridging and onycholysis. At 7 years of age, following informed consent, the patient was enrolled in the Ospedale Pediatrico Bambino Gesù “Undiagnosed Patients Program”, and a trio-based whole exome sequencing analysis, together with a novel skin biopsy, was performed (for methods, see Appendix S1). Variant filtering, annotation and prioritization allowed to identify the de novo c.2T>C (p.Met1?) in KLHL24 (NM_017644) as the only putative disease-causing event. Sanger sequencing confirmed the variant de novo occurrence (Fig. S1). While this work was ongoing, this mutation was reported as causative for a novel form of autosomal dominant EBS (2, 3). Consistently, IFM showed cleavage within the epidermal basal layer and preserved expression of epidermal adhesion proteins, including keratins 5 and 14 (Fig. S2a, b). EM of perilesional skin revealed a decrease in tonofilaments in basal keratinocytes, and examination of additional sections of the biopsy performed at birth showed reduced tonofilaments in basal keratinocytes and focal cytoplasmic vacuolization (Fig. S2c–e).
Fig. 1. Clinical features. Case 1: (a) hypopigmented polymorphic scars on the back, (b) hypopigmented, atrophic, and stellated scars on the calf, and (c) raised stellated scarring intermingled with skin atrophy on hand dorsum at the age of 7 years. Case 2: (d, e) residual skin erosions on the knees, legs and left wrist at 14 days of age, (e) note the hypoplastic nail of the third finger; (f) atrophic scarring, milia, and follicular atrophoderma on the forearm and hand dorsum at 2 months of age.
Case 2. Female, born from healthy parents at 38 weeks of gestation weighing 2,530 g. She presented extensive denuded skin areas on the legs and foot dorsum, wrists and hands, columella and upper lip, requiring hospitalization (Fig. 1d, e). Several finger- and toe-nail plates appeared hypoplastic (Fig. 1e), while mucosae were not affected. IFM of a skin biopsy showed normal expression of epi-thelial adhesion proteins, including keratins 5 and 14, and cleavage within the basal epidermal layer. EM confirmed the basal cleavage (Fig. S2b, f), and also revealed a reduction of tonofilaments in basal keratinocytes. Thus, a diagnosis of EBS was established. In the following weeks, she developed residual atrophic and linear scarring, follicular atrophoderma, and milia, and a few new blisters (Fig. 1f). Mutation analysis for known EB genes was originally negative. Subsequently, sequencing of KLHL24 allowed to identify the heterozygous mutation c.3G>A (p.Met1?) (Fig. S1). Currently, at the age of 5 years, the patient continues to develop a few and small lesions at trauma sites, and presents onychodystrophy and growth delay (weight <third centile).
Case 3. Male, born from healthy parents at 39 weeks of gestation, weighing 2,580 g. He presented large skin defects affecting his limbs and abdomen, which healed rapidly leaving hypopigmented atrophic scars and toenail dystrophy (Fig. S3a, b). Molecular analysis of EB genes revealed the de novo heterozygous c.2T>C change in KLHL24 (Fig. S1).
As cardiac disease has been reported recently in patients with EBS due to KLHL24 mutations (EBS-KLHL24) (2, 7), our children also underwent screening for cardiomyopathy markers, including creatine kinase with muscle band, N-terminal-pro-brain natriuretic peptide, troponin-I, and cardiology examination, which were all normal.
Our data confirm that EBS-KLHL24 is characterized by congenital skin defects of the lower limbs, which heal rapidly, leaving hypopigmented-atrophic patches and peculiar stellate and linear raised scars. However, disease severity at birth is quite variable, as skin denudation may also involve the upper limbs, face and trunk. Skin fragility can markedly improve already within the first weeks of life, as exemplified by cases 1 and 2. Interestingly and in apparent contrast with the minimal residual skin fragility, a persistent reduction in tonofilaments in basal keratinocytes was observed in late childhood in case 1, suggesting that additional factors may contribute to the formation of skin lesions during intrauterine life. Cases 1 and 2 developed follicular atrophoderma and hair loss during infancy to childhood, thus confirming previous reports of hair abnormalities in some patients (2). An intriguing feature common to our patients is late preterm and slightly low weight at birth, persisting until the last follow-up at age 5 years in patient 2. Indeed, reduced growth and low weight were noted in a KLHL24 start codon mutation knock-in mouse model (3). Further studies are needed to ascertain the frequency of these features and the possible relationship with KLHL24 mutations. Indeed, the wide tissue distribution of KLHL24 suggests that mutations could affect other organs, in addition to skin (2). We recently found evidence of dilated cardiomyopathy (DCM) in 8 out of 20 EBS-KLHL24 patients (40%), the youngest being 25 years old (8). One additional EBS-KLHL24 family with lethal DCM has been reported (7). In our cases, cardiological examination and sensitive markers for cardiac dysfunction all proved negative, even though the young age of the patients does not exclude possible future cardiac complications.
Overall, our findings document further the relatively high rate of de novo occurrence of specific KLHL24 nucleotide substitutions, which define a mutation hot-spot that appears to be selected by function. Our 3 Italian patients add to the 2 recently reported familial cases (2), indicating that KLHL24 mutations represent a non-negligible subset of Italian patients with EBS. Early diagnosis is crucial to appropriate genetic counselling and to ensure adequate follow-up and prompt treatment for cutaneous and possible extracutaneous manifestations.
Authors acknowledge the support from the Italian Ministry of Health (Ricerca Corrente 2018-2020).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize