


1Medical Biology Laboratory, 6Biostatistics Department and 7Medical Imaging Department, Claudius Regaud Institute, Toulouse University Cancer Institute (IUCT-O), 2CRCT, Inserm, University of Toulouse III - Paul Sabatier, CNRS, 3University of Toulouse III - Paul Sabatier, 4Thoracic Oncology Department, Larrey Hospital, University Hospital of Toulouse, and 5Dermatology Department, University Hospital of Toulouse, Toulouse University Cancer Institute (IUCT-O), Toulouse, France
Antibodies targeting immune checkpoints were recently approved for metastatic melanoma. However, not all patients will respond to the treatment and some will experience grade III–IV immune-related adverse events. Therefore, early identification of non-responder patients would greatly aid clinical practice. Detection of circulating tumour DNA (ctDNA) is a non-invasive approach to monitor tumour response. Digital droplet PCR was used to quantify BRAF and NRAS mutations in the plasma of patients with metastatic melanoma treated with immunotherapy. In 16 patients, ctDNA variations mirrored tumour response (p = 0.034) and ctDNA augmentation during follow-up detected tumour progression with 100% specificity. In 13 patients, early ctDNA variation was associated with clinician decision at first evaluation (p = 0.0046), and early ctDNA increase with shorter progression-free survival (median 21 vs. 145 days; p = 0.001). Monitoring ctDNA variations early during immunotherapy may help clinicians rapidly to discriminate non-responder patients, allow early adaptation of therapeutic strategies, and reduce exposure to ineffective, expensive treatment.
Key words: ctDNA; melanoma; immunotherapy; biomarker; therapeutic response.
Accepted Nov 1, 2018; Epub ahead of print Nov 5, 2018
Acta Derm Venereol 2019; 99: XX–XX.
Corr: Dr Anne Pradines, Medical Biology Laboratory, Claudius Regaud Institute, Toulouse University Cancer Institute (IUCT-O), FR-31059 Toulouse Cedex 9, and Pr. Nicolas Meyer, CRCT, Inserm, University of Toulouse III - Paul Sabatier, CNRS, FR-31000 Toulouse Cedex 9, France. E-mail: pradines.anne@iuct-oncopole.fr; meyer.n@chu-toulouse.fr
Immunotherapy has yielded a dramatic improvement in the prognosis and survival of patients with unresectable melanoma. However, not all patients respond to treatment and some experience severe adverse events. The detection of mutations in peripheral blood (i.e. ctDNA) is a new, non-invasive approach to monitor tumour response. Using a sensitive method, this study showed that early assessment of ctDNA variation during the course of therapy could predict the tumour response to treatment. These results may help clinicians to rapidly discriminate non-responders, allowing early adaptation of therapeutic strategies and reducing their exposure to ineffective and costly treatment.
Immune checkpoint inhibitors (anti-CTLA-4, anti-PD1 or combination of both) have revolutionized the treat-ment of metastatic melanoma (1) with striking results on overall survival (OS) in advanced-stage metastatic melanoma (the 3-year OS were, respectively, 34%, 52% and 58%) (2). However, this mode of treatment is effective only in a subset of patients as 40–65% have shown minimal or no RECIST (Response Evaluation Criteria in Solid Tumours) response and 43% of responders develop acquired resistance by 3 years (2). In addition, up to 55% of patients may experience grade 3–4 immune-related adverse events (2–5). Today, none of predictive biomarkers appear sufficiently robust and easily transferable into clinical practice to guide treatment choices. Indeed, intratumoural lymphoid infiltrates and tumoural PD-L1 expression at baseline both have a lack of discriminative capacity between responders and non-responders (6, 7).
The interpretation of PD-L1 immunostaining on primary tumour biopsies is currently limited by spatiotemporal intratumoural heterogeneity. As a consequence, the choice of treatment and evaluation of its efficacy is based on clinical–radiological criteria 12 weeks after induction, since contrary to chemotherapies, immunotherapy (IT) demonstrates a delayed response to treatment. Thus, development of new, early prognostic and predictive biomarkers to enable fine monitoring of tumour response is critical, as it may: (i) reduce exposure to potentially toxic and expensive treatments in non-responders; (ii) allow for early adaptation of therapeutic strategies in non-responders; and (iii) provide useful information for the management of adverse events in responders.
Detection of mutations in circulating cell-free tumour DNA (i.e. ctDNA) is a useful, non-invasive and repeatable approach that can be used to assess the mutational status of tumours in different cancers (8). ctDNA has been correlated with baseline tumour burden (9–11) and with tumour-size evolution (12). Moreover, it overcomes tumour heterogeneity. Our team has recently shown the utility of detection of BRAFV600E and KRAS mutations in plasma to monitor non-small-cell lung cancer evolution (13, 14) and to discriminate pseudo-progression cases (15).
In melanoma, detection of BRAFV600E in ctDNA has been explored in various studies and correlated with clinical outcomes in patients treated with BRAF and MEK inhibitors (16–19). Serial analysis of ctDNA can identify the genomic evolution of metastasis in response to targeted therapy (16). More recently ctDNA has also been suggested to provide information on therapeutic responses to immunotherapy in patients with melanoma (12, 20). However, these seminal studies explored ctDNA detection rather belatedly, 8 weeks after the first infusion. A crucial objective is to identify as early as possible those patients who will never respond to immunotherapy.
We report here a retrospective analysis of ctDNA assessed by digital droplet PCR (ddPCR) in 22 patients undergoing immunotherapy for metastatic melanoma. This study aimed to determine whether ctDNA variations or interpretation of early changes were associated with tumour response to immunotherapy.
From February 2014 to November 2016, 22 patients with stage IIIC–IV melanoma with BRAF and NRAS mutations, detected by high-resolution melting and TaqMan assay when a variant was detected in archived formalin-fixed, paraffin-embedded tumour specimens, and treated with immunotherapy at our institution, were included in our cohort. In our protocol, patients had been sampled at baseline and before each infusion of treatment. All patients gave their informed consent to participate in this study and the local ethics committee approved the study (DC-2009-989).
Patients were treated with immune checkpoint inhibitors: pembrolizumab (200 mg every 3 weeks), nivolumab (3 mg/kg every 2 weeks in monotherapy), or ipilimumab (3 mg/kg every 3 weeks in monotherapy) or a combination of both drugs (respectively 1 and 3 mg/kg every 3 weeks) (see Table SI for therapeutic schedules).
Tumour imaging was performed with computed tomography, positron-emission tomography, and/or magnetic-resonance imaging, as appropriate, every 12 weeks. Therapeutic response was assessed using RECIST 1.1 criteria (21) and a physician interpreted each case. Stable disease was confirmed on further tumour evaluation.
Before each infusion, two 7-ml blood samples were taken from each patient in ethylenediaminetetraacetic acid (EDTA) tubes (BD Biosciences) and double centrifuged (10 min 1,200 g, supernatant collected then centrifuged again for 10 min 16,000 g) to isolate the plasma. Samples were stored at –80°C within 4 h of collection. Cell-free DNA (cfDNA) was purified from 2 ml plasma using the QIAmp circulating nucleic-acid kit according to manufacturer’s recommendations (Qiagen). ctDNA was tested by ddPCR for the presence of corresponding BRAF (V600E or V600K) or NRAS mutations (Q61K, Q61R, Q61L, Q61H) with specific paired PrimePCR™ ddPCRTM Mutation Detection Assays (Bio-Rad) on QX200 system (Bio-Rad). Input DNA was emulsified into 20,000 droplets, amplified by PCR, fluorescent labelled, and then read with a QX200 Droplet Reader using QuantaSoft (Bio-Rad). For each mutation, the false-positive rate was estimated with plasma containing only WT DNA at similar concentrations as those found in patients, in triplicate.
We defined “variation” of ctDNA concentration in accordance with the following formula: ΔctDNA=((ctDNA)n+1–(ctDNA)n)/(ctDNAn) and considered as significant ctDNA variation any ctDNA concentration changes greater than 20% between 2 assessments.
For correlation of ctDNA variations and radiological tumour evaluation, image data and ctDNA variations of 16 patients were summarized as binary covariates, progression or non-progression (gathering objective response or stable disease) and increase or decrease between 2 visits, respectively.
For early ctDNA variation, increase or decrease was assessed using ctDNA at baseline as reference in only 13 patients. Time to progression (TTP) was calculated from the second blood draw until the first clinical–radiological confirmation of tumour progression. Progression rates were estimated by the Kaplan–Meier method, with 95% confidence intervals (CI). Univariate analyses were performed using the log-rank test.
Associations between 2 qualitative covariates were assessed using Fisher’s exact test. Two-sided p-values of < 0.05 were considered statistically significant. All statistical analyses were performed using STATA 13.0 software (STATAcorp, College Station, CA, USA).
ctDNA was analysed in 122 plasma samples from 22 patients with melanoma. Median follow-up was 135 days (range 20–620 days). Patients harboured a BRAF or NRAS mutation, mostly stage IV, and 68% had received a prior line of systemic therapy (chemotherapy or targeted therapy, immunotherapy) before a new line of treatment with immunotherapy (Table I).
Table I. Characteristics of patients assessed for progression-free survival (n = 13)
The mutation initially identified in the primary tumour was detectable in the ctDNA of 21/22 (95%) patients in at least one sample during immunotherapy treatment (Fig. S1). For 1 patient, the mutation of interest in ctDNA was totally undetectable at baseline and during follow-up; however, the patient had previously been determined to have NRASQ61L mutated, according to high-resolution melt-screening, for BRAF, NRAS, CKIT mutations on a single metastatic adenopathy that had been diagnosed 3 years previously.
Examples of ctDNA monitoring in a non-responder patient, a patient sampled at progression, and a patient exhibiting a long-term response are shown in Fig. 1.
Fig. 1. Three examples of ctDNA monitoring immunotherapy courses. Patient number 21 bearing a metastatic BRAFV600E mutated melanoma was treated with a combination of anti-PD-1 and anti-CTLA-4 after a relapse following BRAF and MEK inhibitors (nodal, lung and adrenal metastases). First radiological evaluation at day 98 showed dramatic evolution of the metastases. BRAFV600E was undetectable at baseline, then increased significantly after the third cycle (day 58). Lactate dehydrogenase (LDH) (reference values 125–220 UI/l) detected tumour growth later than ctDNA. The first positron-emission tomography (PET) scan evaluation (day 119) of patient number 7 (NRASQ61K mutated) treated with anti-PD-1 showed a partial response with decrease in size of lung and peritoneum metastases, while the second PET scan (day 224) revealed a progressive disease. ctDNA evolution paralleled tumour evolution, whereas LDH (reference values 87–241 UI/l) remained stable during disease evolution. Patient number 13 presented pulmonary and hepatic secondary lesions of a NRASQ61L mutated melanoma at initiation of antiPD1 treatment. First scan evaluation (day 126) reported a complete response of pulmonary lesions and a dramatic decrease in hepatic metastases, with a much more necrotic aspect. Complete ctDNA clearing was observed as soon as the second cycle (day 21). As the patient developed an interstitial nephropathy at the 5th cycle, treatment was discontinued and the patient was placed under active surveillance. No relapses were observed during follow-up and ctDNA remained totally undetectable. LDH (reference values 125–220 UI/l) presented 2 peaks at 1.5× normal upper limit. The second peak might reflect nephropathy, and LDH is not in this case contributive to the assessment of tumour evolution.
We then tested associations of variations of ctDNA concentration (see Methods) with radiological tumour-assessment between 2 consecutive visits during patient follow-up. Our analyses were performed only on patients presenting with measurable ctDNA (n = 16) and samples that were concomitant with medical imaging (median delay between samples and imaging: 16.5 days, min–max: 0–46; Fig. S1). Minimal ctDNA variation observed in enrolled patients was 21.3%. We found that an increase in ctDNA detected progressive disease with 63% sensitivity (n = 7/11) and 100% specificity (n = 5/5) (Table II). ctDNA diminution did not detect progression with a probability of 0.555 (n = 5/9). Variation in ctDNA during the follow-up was associated with the imaging results (p = 0.034; Table II).
Table II. Circulating tumour DNA (ctDNA) variation and clinical conclusions between 2 consecutive imaging during patients follow-up (n = 16)
For 15 patients, the first plasma sample was collected at baseline (Fig. S1) and ctDNA was detected in 11/15 (73%) patients. Since our retrospective study evaluated the prognostic value of early ctDNA variation, the analysis was conducted on only 13 patients with baseline and early (before the second or third treatment infusion) blood samples available (Table SI and Table SII and Fig. S2). The median delay between the 2 consecutive blood draws was 26 days (17–73). Minimal variation of ctDNA concentration observed in enrolled patients was 25%.
Patients were categorized according to therapeutic response. Six patients were considered primary non-responders (non-equivocal progression leading to a therapeutic switch at first evaluation) and 7 patients were considered responders, according to RECIST 1.1 criteria (21). Early ctDNA-positive variation detected all responder patients and early ctDNA-negative variation detected 5 out of 6 responders. A significant association was found between early ctDNA variation and tumour response at the first evaluation (p = 0.0046, Table III, Table SII and Fig. S2).
Table III. Early circulating tumour DNA (ctDNA) variation and clinical outcome of patients at first scan evaluation (n = 13)
It was then analysed whether this ctDNA variation from baseline was associated with progression-free survival (PFS). The median PFS was 21 days for patients with increased ctDNA vs. 145 days for patients with decreased ctDNA. PFS was significantly better for patients with decreased ctDNA (p = 0.001; Fig. 2).
Fig. 2. Kaplan–Meier curve of progression-free survival (PFS) according to circulating tumour DNA (ctDNA) early variation groups. Progression rates were estimated in 13 patients using the Kaplan–Meier method with 95% confidence intervals (95% CI). Black: increased ctDNA (n = 5); Grey: decreased ctDNA (n = 8). Log-rank test: p = 0.001.
Detection of ctDNA by ddPCR has been widely assessed in patients with melanoma treated with targeted therapy or immunotherapy (18, 19, 22). It is an accurate, highly sensitive and reproducible technology that provides absolute quantification of circulating mutations (23). It has been found to be a perfectly adapted technology to monitor tumour evolution once the driver mutation has been first identified in tumour biopsy. In our cohort, we observed the detection rate of mutations in ctDNA at baseline (73%), similar to that reported previously (73% in Gray et al. (17), 84.3% in Sanmamed et al. (18), 76–81% in Santiago-Walker et al. (19), 73% in Lee et al. (20)).
A major conclusion of the current study is that if an increase in ctDNA is associated with tumour progression, a decrease in ctDNA should be subject to clinical-radiological evaluation. We identified 4 “discordant” situations in which ctDNA change was negative, whereas patients experienced progressive disease. First, exclusive intracranial metastases are reported to shed very small amounts of DNA in peripheral blood; thus, in this case, cerebrospinal fluid could be a good surrogate for ctDNA (24). Secondly, ctDNA interpretation can also be hampered in cases of multiple metastasis with dissociated response. Indeed, this patient showed a progressive disease with dramatic increase in hepatic metastases and apparition of subcutaneous metastases despite diminution of pulmonary and vertebral lesions. This case presents issues with the origin of ctDNA release that cannot be identified precisely and may vary from one metastasis to another. Then, we hypothesize that clonal heterogeneity may have induced false-negative results in the 2 other discordant patients with the emergence of new clones that had distinct mutations from the one we targeted (25). This limitation may be overcome by broader characterization of the genome (12, 16) with next-generation sequencing technologies to allow identification of the genetic landscape of tumour resistance, generated by selection pressure of the treatment (26).
As ctDNA-positive variation during treatment could discriminate tumour progression with high specificity, this raised the question, like a proof-of-concept, as to whether such variation, assessed before the first clinical and radiological evaluation, could predict patients who will not respond to the treatment. A positive ctDNA variation from baseline was associated with a therapeutic change decision at first evaluation and was also associated with lower progression-free survival. These results need first to be confirmed on a larger and independent cohort to comply with REMARK guidelines. Nevertheless, they also corroborate Lee et al.’s (20) and Cabel et al.’s (12) previous studies in which undetectability of ctDNA during the course of the treatment was associated with better PFS (12, 20). The current study revealed other aspects that it would be interesting to confirm using a validation cohort study: (i) considering ctDNA variations and not solely ctDNA detectability would allow to discriminate every patient, suggesting that patients with a decreased, but still detectable, ctDNA would be considered as responders; (ii) it would be possible to be aware of tumour response as soon as the second infusion of the treatment is applied. Therefore, observing an early positive variation could become a reliable and helpful prognostic biomarker for clinicians in daily practice, allowing rapid treatment adaptation that could improve patient prognosis, reduce patient exposure to an ineffective therapy, and decrease the prescription of expensive treatments.
This proof-of-concept study outlines the usefulness of ctDNA variations to monitor patients treated with new immune-checkpoint blockers. In order to implement ctDNA monitoring in routine clinical decision-making, further research is needed to compare ctDNA with medical imaging in a dedicated assay in which clinician decisions rely on ctDNA, also taking into account the medico-economic viewpoint.
The authors thank the Société Française de Dermatologie and the Agence Nationale de la Recherche (ANR-10-NANB-0002) for their financial support.


 Click to show fullsize
Click to show fullsize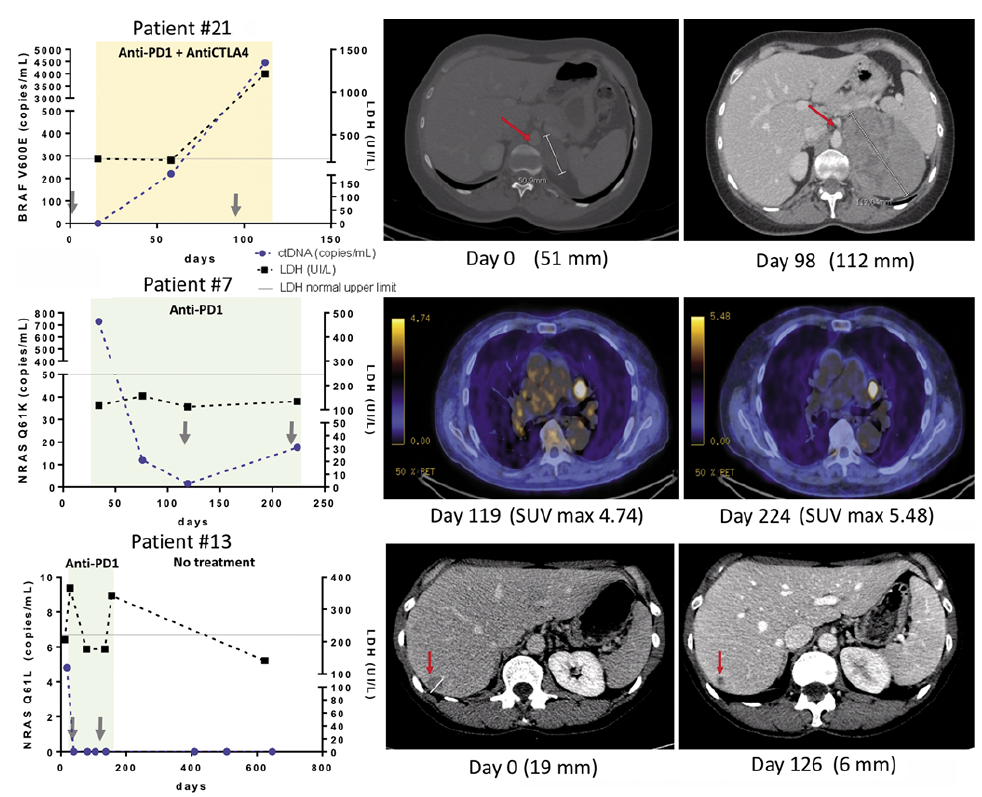 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize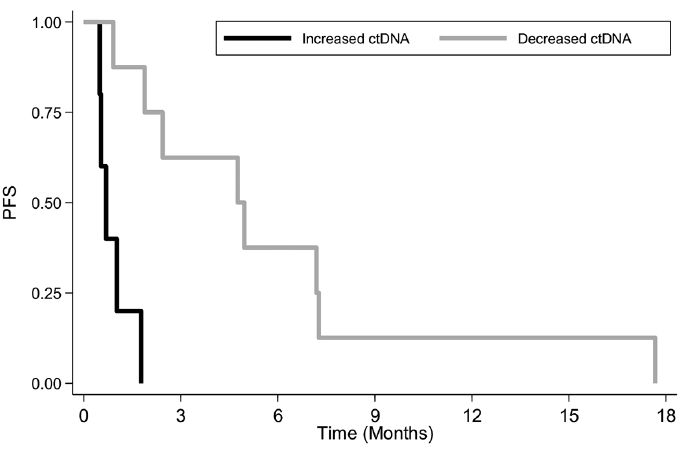 Click to show fullsize
Click to show fullsize