


1Department of Dermatology, Severance Hospital, Cutaneous Biology Research Institute, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, and 2Department of Pediatrics, Severance Children’s Hospital, Seoul, Korea. *E-mail: oddung93@yuhs.ac
Accepted Nov 20, 2018; Epub ahead of print Nov 21, 2018
Aplasia cutis congenita (ACC) is a rare congenital disorder characterized by focal, localized absence of skin or defects of the skin in neonates. The estimated incidence of ACC is approximately 0.5–1 in 10,000 newborns (1). The clinical appearance of ACC is heterogeneous and it can be associated with various systemic congenital anomalies. However, the causative gene mutation is unclear. Frieden (2) proposed a classification of ACC based on clinical characteristics, such as involved body area, associated anomalies, and inheritance patterns. Most cases of ACC manifest as a solitary lesion involving the scalp, especially at the vertex. However, ACC is also associated with underlying malformations, such as limb or skull defects, or organ involvements, including central nervous, cardiovascular, and gastrointestinal systems (1–4). ACC treatment is controversial, but most cases heal spontaneously within a few weeks with conservative treatment (1, 3, 4).
We report here a rare presentation of ACC with multifocal, symmetrical, discrete skin lesions over the entire body, including the scalp and extremities, in a newborn male.
A full-term male infant born at 39 weeks of gestation (birthweight 2,560 g) was transferred to the neonatal intensive care unit (NICU) with multifocal full-thickness skin defects over the entire body: scalp, trunk, and extremities. His parents denied any family history of congenital skin disease, and there was no history of infectious disease, trauma, and maternal intake of drugs in the prenatal period. The mother had a history of one previous pregnancy, resulting in a normal child. During prenatal evaluation, no signs of systemic or macroscopic placental anomaly were detected.
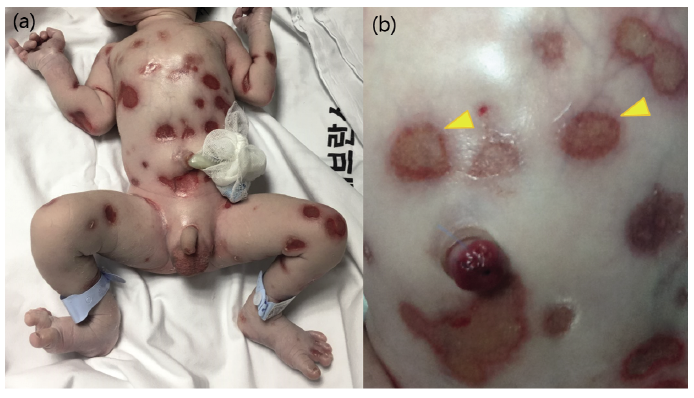
At delivery, more than 30 individually demarcated, well-defined, coin-sized, full-thickness skin defects were noted on the entire body, including the scalp and extremities (Fig. 1). However, there was no sign of blister formation or mucosal erosions, and the distal ends of the patient’s digits were intact with no dystrophy of the nail units. The patient underwent a thorough evaluation, including skull and whole-spine radiography, brain ultrasonography, and echocardiography, to detect possible anomalies, and there were no additional systemic abnormalities apart from the skin manifestations. A genetic assay panel for possible dysmorphisms (ARX, FOXC2, PLEC, LAD1, etc.) was performed using next-generation sequencing combined with direct sequencing. No genetic mutations associated with congenital bullous disease were noted.
Fig. 1. (a) Full-thickness skin defects immediately after birth. (b) Within a few hours, several lesions covered with thin yellowish crusts with signs of spontaneous healing (arrowhead).
Since no newly formed lesions were observed during the first few days after birth and several lesions showed signs of spontaneous healing, it was decided to provide conservative treatment with no additional surgical treatment. The infant was placed in incubator care with a humidity of more than 80%, in order to prevent excessive trans-epidermal water loss and desiccation of healing lesions. He was treated with semi-occlusive dressings twice a day for the first week after birth. To prevent maceration due to wound exudates, a combination mixture of petrolatum ointment, paraffin emollient and mupirocin ointment was used, covered with antibacterial dressing material (Sorbact® Compress, ABIGO Medical AB, Sweden). The dressing protocol was continued for the following weeks with occasional wound swabs to detect any possible infection. Prophylactic antibiotics (ampicillin/sulbactam, cefotaxime) were initiated, but were discontinued after 10 days when subsequent swab culture results showed no bacterial growth. After 3 weeks, the patient was discharged from the NICU when more than 70% of the lesions showed re-epithelization. Some lesions, especially on the flanks where the defects were most extensive at birth, presented with peripheral cicatrization, but the surrounding skin did not show contracture.
Until the age of 12 months, no medical problems were apparent, and the infant showed completely normal development on routine paediatric examination. No new lesions developed, most lesions were fully epithelized, and visible hair follicles were observed from healed lesions on the scalp (Fig. 2).
Fig. 2. (a) Well-healed skin defect leaving whitish scars without contracture at the age of 9 months. (b) Hair regrowth on healed scalp lesions (circle).
This case involved atypical manifestation of ACC confined to the skin with no congenital defects or systemic organ anomaly. The case presented extensive involvement of aplasia skin lesions, similar to type 5 ACC, which is defined by Friden (2) as ACC associated with foetus papyraceous or placental infarcts. Most cases of ACC with foetus papyraceous have been presented in association with loss of 1 foetus during a monochorionic twin pregnancy (5–7). However, the affected foetus was not associated with foetal demise of a twin or macroscopic placenta abnormalities. Thus, we consider this patient as having type 5 ACC without foetus papyraceous or placental abnormalities.
The frequency of multiple skin defects in ACC is extremely rare; one retrospective study showed only 3 cases (3.3%) of multiple (5 or more) cutaneous lesions in a total of 90 cases of ACC (8), while another study reported no multiple lesions in total of 22 cases of ACC (9). In addition, the majority of cases of ACC appear as isolated cutaneous lesions, but skull defects are found in approximately 30% of cases (8–10).
It is important to consider congenital epidermolysis bullosa (EB) as a differential diagnosis. EB is characterized by extremely fragile skin, widespread bullae, and erosions after subtle mechanical trauma (3). EB can be distinguished from our case based on its chronic course with recurrent relapse and discovery of gene defects through genetic study, as our patient did not show any recurrence since birth. In addition, there are only a few case reports on symmetric truncal ACC without congenital anomalies, but none of these cases showed numerous, discrete, well-defined skin defects as observed in this case (7, 9, 11).
The size, location of the lesions and associated complications should be considered, in order to determine the treatment plan for ACC. A review of the literature revealed that superficial, small areas of ACC (less than 2–3 cm diameter) are usually well healed with conservative wound care, such as petrolatum, bacitracin, silver sulphadiazine or occlusive dressings (1, 3, 12). Surgical options, such as skin grafts, local flaps or tissue expansions, are considered in cases of larger defects of skin and underlying bone. Furthermore, arising complications, such as infection, haemorrhage, or significant electrolyte imbalances, may require surgical correction (1, 3, 4, 6). However, even if the lesions are multiple and extensive, conservative methods were sufficient for successful re-epithelization if there were no complications related to ACC (4, 7, 13).
The current case had unique clinical features, including multifocal full-thickness skin defects with extensive, symmetrical involvement without other associated systemic involvements. It is notable that, despite the extensive skin lesions, most of the defects healed spontaneously with conservative care and observation during the early days of life, with no additional surgical procedures, implying an excellent clinical prognosis for the disease.


 Click to show fullsize
Click to show fullsize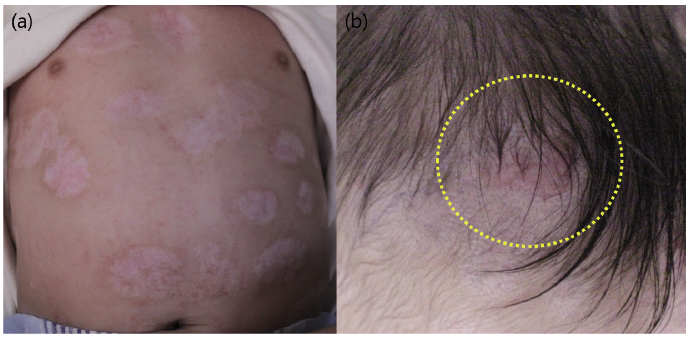 Click to show fullsize
Click to show fullsize