


1Dermatology Unit, 2Department of Pathology, and 3Department of Paediatric Haematology-Oncology, Bambino Gesù Children’s Hospital, Piazza Sant’Onofrio, 4, IT-00165 Rome, Italy. E-mail: roberta.rotunno@opbg.net
Accepted Feb 26, 2019; E-published Feb 27, 2019
Langerhans cell histiocytosis (LCH) is a rare childhood disease of the monocyte-macrophage system characterized by a clonal, uncontrolled proliferation and accumulation of CD1a+/ CD207+ dendritic cells (DCs). LCH is stratified into single system disease (unifocal or multifocal), multiorgan disease without organ dysfunction, and multiorgan disease with organ dysfunction. The clinical presentation ranges from isolated, self-healing skin and bone lesions, to life-threatening multi-system disease, and the symptomatology is extremely wide. Thus, the course, treatment and prognosis vary according to LCH type. Skin lesions represent the second most-common clinical manifestation of LCH (30–60%). The eruption may involve the scalp, intertriginous area, face, trunk and buttocks. Cutaneous presentations are polymorphic, from erythematous, yellow scaly or crusted papules, to macerated patches, pustules, vesicles, or petechiae and purpura (1, 2).
We report here the case of a girl, in remission from LCH, with subsequent appearance of agminated spitzoid naevi on the neck, and in the axillae, inguinal folds and vulva.
This 6-year-old Caucasian girl, followed for LCH in remission, was referred to the Dermatology Department for the annual skin check and for evaluation of her flexure freckling.
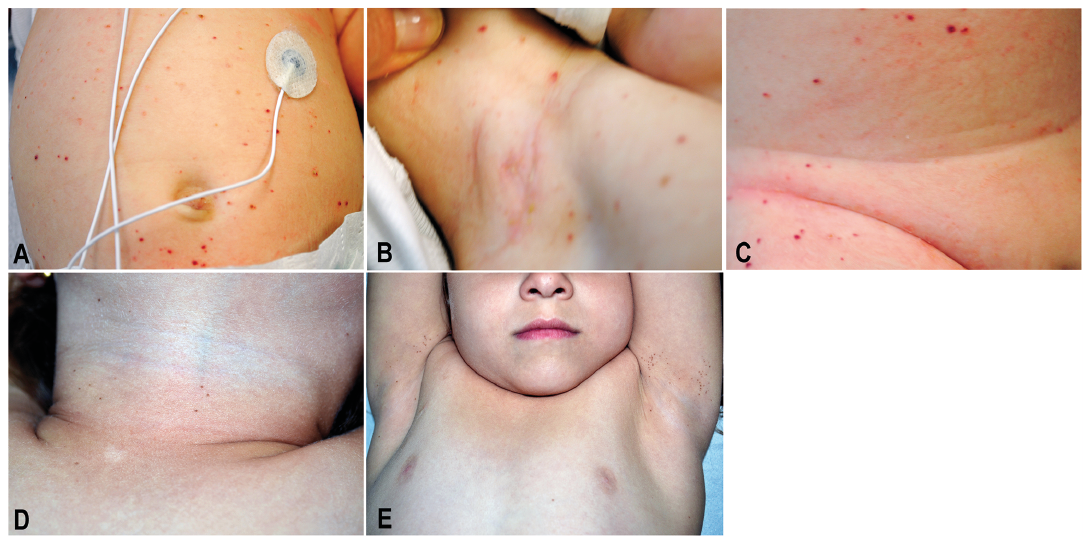
LCH had been diagnosed by skin biopsy when the child was 2 months old (Fig. S1 A, B). On the first admission, she arrived with fever, hepatosplenomegaly and anaemia, and presented with diffuse papular and purpuric lesions on the trunk and inguinal and axillary folds (Fig. 1 A–C), and crusted yellow papules on the scalp. No bone marrow involvement was detected.
She was treated with vinblastine (6 mg/m2 i.v.), oral prednisone (40 mg/m2 daily) and methotrexate (500 mg/m2 24 h-infusion with folinic acid rescue), because of skin, liver and spleen involvement with complete regression within two years. At 5 years of age, she began to develop freckling on the neck and bilateral axillary and inguinal regions. Physical examination revealed several clustered brown macules, 2 to 4 mm in diameter, on the neck, bilateral axillary and inguinal folds, and external genitalia (Fig. 1 D, E). The differential diagnosis included lentigines, naevi or postinflammatory melanosis. On dermoscopy these pigmented lesions showed a reticular pattern with sporadic globules. Finally, a biopsy of two macules was performed and histologic examination revealed junctional spitzoid naevi consisting of nests of melanocytes with epithelioid morphology at the dermo–epidermal junction; immunohistochemical staining for B-RAF V600E mutant protein was negative in nevus cells (Fig. S1 C, D).
Fig. 1. Numerous diffuse crusted or scaly papules and purpuric lesions on the trunk (A), axillary folds (B) and inguinal (C). Several clustered brown macules, 2 to 4 mm in diameter, were present on the neck (D) and bilaterally on axillary (E) and inguinal folds (not shown).
To our knowledge, a correlation between LCH and eruptive agminated naevi has been described in 4 other children (3–5). Our case is the first Italian patient reported in the literature. There are several similarities between the cases suggesting a potential causal and not coincidental association between LCH and eruptive naevi. LCH skin involvement was described in 4 of 5 patients; however, all patients presented pigmented lesions in the folds, especially the groin and axillae. None of the patients developed naevi on the scalp, the area mostly involved in LCH (3–5). In 3 cases the lesions were diagnosed as Spitz naevi or junctional naevi with spitzoid features (3, 4); Feldstein et al. (5) reported a diagnosis consistent with agminated junctional naevi. In another case no biopsy was performed due to clinical features of benign melanocytic lesions (4). Our histological examination revealed features of junctional naevi, with spitzoid morphology.
Four patients were treated with chemotherapy for systemic involvement (3, 4). Surinach et al. (4) reported two cases treated with vinblastine and prednisone, one of them also being treated with topical mechlorethamine. One patient received only vinblastine (3). The case described by Feldstein et al. (5) was treated with topical corticosteroids only. Our case received vinblastine, prednisone and methotrexate treatment (Table SI).
Pathogenesis of agminated naevi in patients with LCH history is not yet entirely clear. Eruptive naevi can be idiopathic or linked to trauma, burns, cutaneous mastocytosis, and primary adrenocortical insufficiency (6–8). In the literature, the development of multiple melanocytic naevi is often associated with an underlying trigger (6). It has been commonly associated to severe blistering skin diseases, such as naevi in epidermolysis bullosa, or to immunosuppressive conditions (7–10). Relationship with various pharmacologic treatments (i.e. azathioprine, corticosteroids, combination chemotherapy) has already been described (11). Recently, the common use of biologics has become another possible cause of eruptive naevi (10, 11). According to some reports on LCH and agminated naevi, chemotherapy might play a role, but was not administered in one of the reported patients. Despite the appearance of melanocytic naevi on areas previously affected by LCH, Berk & Lane (3) postulated that this manifestation was coincidental. Later, Surinach et al. (4) and then Feldstein et al. (5) suggested an immune-mediated mechanism with local immune dysregulation and inflammation. They also considered LCH to be an additional cause of eruptive melanocytic naevi, characterized by a specific distribution tracing some of the previous LCH cutaneous lesions. This could be justified by the immune tolerance induced in LCH lesions by high levels of regulatory T cells (CD4 CD25 FoxP3 T-regs) (5, 12). Another explanation could be that the proinflammatory cytokine cascade secreted in LCH lesions, especially IL-1, known to upregulate melanocortin-1 receptor (MC1R), may play a role in naevogenesis, stimulating melanocytic lesion development (5, 12). However, the timing of the naevi eruption and whether these naevi are developed de novo or by subclinical nevus nests is still unknown. In all the described cases this event occurs several years after LCH remission and when cytokine levels are presumably normal (3–5). The absence of naevi on the scalp in all reported patients has yet to be explained. In our opinion, local trauma could explain the localization of the naevi on the folds and other frictional areas, even when cutaneous LCH involved the whole body. Damaged keratinocytes and local cellular interactions can influence melanocyte growth. Inflammatory cells can release growth factors inducing naevogenesis, and immunosuppression could stop the cellular surveillance keeping preexisting naevus cell nests in check. Thus, in predisposed patients, the specific trigger factor for the development of eruptive naevi could be frequent and localized friction (6). Considering the hypothesis of a common origin of epidermal melanocytes and Langerhans cells from epidermal cambial cells, we searched for the B-RAF mutation in both neoplastic Langerhans cells and naevocytic melanocytes (13). Indeed, B-RAF V600E mutant protein is considered a driver mutation in a proportion (≈55%) of LCH patients with activation of the MAPKinase RAS-RAF-MEK-ERK cell signalling pathway. Although 45% do not have BRAF V600E mutation, ERK was reported to be activated in pathologic histiocytes (14, 15). These genetic mechanisms could be assumed to be a common denominator with LCH-associated nevi. However, the molecular study in our patient showed B-RAF positivity in LCH lesions and negativity in melanocytes (Fig. S1 B, D).
In conclusion, LCH could be an additional cause of eruptive melanocytic naevi. However, this should be confirmed in a larger paediatric LCH population, and the pathogenesis should be clarified.


 Click to show fullsize
Click to show fullsize