


Department of Dermatology, The Second Xiangya Hospital, Central South University, Changsha, 410011, Hunan, China. *E-mails: xiaorong65@csu.edu.cn, misseven69@csu.edu.cn
Accepted Mar 21, 2019: E-published Mar 21, 2019
Hyper IgE syndrome (HIES) is a rare primary immunodeficiency disease. Most cases of HIES occur early in life. Nondescript symptoms lead to either misdiagnosis or a delay in diagnosis. We report a case of a child with hyper IgE syndrome who initially presented with recurrent head abscesses and was misdiagnosed as suffering from “folliculitis”.
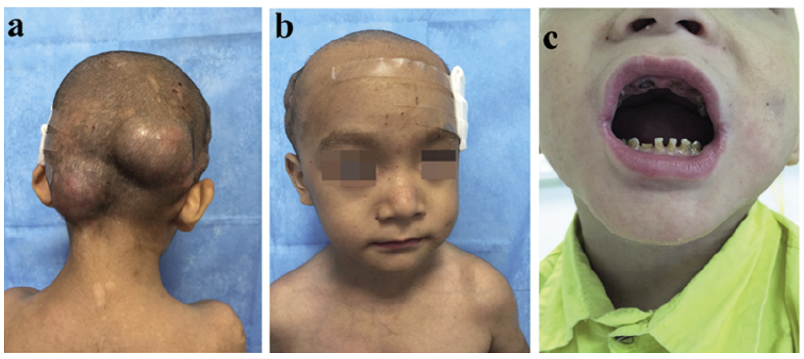
A 5-year-old child was admitted to our hospital with a 4-year history of recurrent head abscesses and aggravation for 1 month. He had been clinically diagnosed with folliculitis and treated with antibiotics, with minimal relief. One month prior to admission, his condition had taken a turn for the worse when several scattered cold abscesses each grew to the size of a fist (Fig. 1a). Further questioning revealed that the patient had a past medical history of repeated eczema, frequent upper respiratory infections (> 10 times/year), and one hospitalization due to supposed septicemia. He had no family history of HIES or any other immunodeficiency disease. There was a characteristic appearance with forehead carina, widely-spaced eyes, and a broad nasal ridge. Other significant physical findings included residual root and tooth defects of the deciduous teeth in the upper and lower jaw, as well as mild lumbar scoliosis (Fig. 1b, c).
Fig. 1. a. Fist-sized abscesses; b. Forehead carina, widely-spaced eyes, broad nasal bridge; c. Residual root and tooth defects of deciduous teeth in the upper and lower jaw.
Laboratory analysis revealed serum IgE levels > 6,000 ng/ml (0–691.4 ng/ml), with an ESR of 29 mm/h (0–15 mm/h) and C-reactive protein of 6.13 mg/l (0–3 mg/l). Routine blood tests revealed a white blood cell count of 14.47 × 109/l (5.0–12.0 × 109/l), a neutrophil count of 8.07 × 109/l (1.8–6.3 ×109/l), an eosinophil count of 1.88 × 109/l (0.02–0.52 × 109/l), and an eosinophil ratio of 13.00% (0.5–5.0 × 109/l). Staphylococcus aureus was detected in the patient’s abscesses. The lumbar plains showed slight scoliosis and the oral plains suggested a high arched palate. The pathological section showed that mixed inflammatory cells, including eosinophils and neutrophils, had infiltrated into the subcutaneous tissue, with associated abscess formation, PAS (–) (Fig. 2a). Urine routine, stool routine, chest X-ray, bone density, and T-cell subsets were all within normal limits.
Fig. 2. Pathologic section: mixed inflammatory cells including eosinophils and neutrophils that had infiltrated into the subcutaneous tissue, with associated abscess formation (hematoxylin–eosin; original magnification: a) × 40; b) × 200).
In 1999, the National Institutes of Health established HIES diagnostic criteria (1) (Table I). In this case, clinical evidence, together with laboratory and histopathological findings, clearly indicated a diagnosis of hyper IgE syndrome (total score: 42 points; see Table I). Significant improvement was noted in the patient following vancomycin and fusidic acid ointment as systematic and topical treatments, respectively.
Table I. NIH-HIES scoring system and patient scores
HIES is a rare disease, with an annual incidence ranging from 1 in 500,000 to 1 in 100,000 (2). There are 2 distinct forms of HIES: Type 1, autosomal-dominant HIES (AD-HIES), is negatively correlated with mutations in STAT3, which is the most prevalent mutation described and accounts for the majority of HIES cases; Type 2, autosomal-recessive HIES (AR-HIES), is mainly caused by dysregulation in DOCK8, TYK2, PGM3, and SPINK5 (3).
HIES is a multisystem disorder associated with varied clinical manifestations. An eczematoid rash is usually the initial clinical manifestation of HIES, generally starting on the scalp and face (4). Recurrent skin infection with S. aureus results in “cold” abscesses lacking the usual features of warmth and erythema, and is a nearly universal feature of HIES. Sinopulmonary infections caused by S. aureus are also common in HIES. A total of 97% of patients have serum IgE > 2,000 ng/ml, and 83% of HIES patients exhibit typical facial features that include a prominent forehead, facial asymmetry, sunken eyes, broad nasal ridge and fleshy nose, prognathism, and craniosynostosis. Approximately 65% of HIES patients have musculoskeletal abnormalities, including hyperextensibility of the joints, scoliosis, osteopenia, and pathological fractures (5). The risk of malignancies, especially lymphoma, should not be overlooked in patients with HIES (6). Cutis verticis gyrata has also been reported in HIES patients (7).
For patients suspected to have hyper IgE syndrome, other immunodeficiency diseases such as Omenn syndrome (OS) and Wiskott-Aldrich Syndrome (WAS), both of which are associated with elevated IgE and eosinophils, should first be excluded. OS is typically manifested by severe early postnatal infection and erythroderma, as well as liver, spleen, and lymph node enlargement, and presents significant cellular immunological dysfunction upon primary immunological screening. Children with WAS experience relatively early onset of symptoms including the triad consisting of eczema, thrombocytopenia, and immunodeficiency.
The therapeutic mainstay during the early stages of hyper IgE syndrome is preventing recurrent infections and subsequent lung remodeling. Treatment with prophylactic antimicrobials effectively eliminates S. aureus and decreases the frequency of pneumonia. Appropriate skin care such as bleach baths or other antiseptic treatments can be undertaken in order to prevent skin infections. HIES patients usually do not need to take precautions to prevent potential fungal infections (4). The role of bone marrow transplantation in HIES remains unclear, although it has been reported that the immunological and non-immunological manifestations of HIES in 2 patients completely disappeared following bone marrow transplantation (8). INF-γ, plasma transfusion, and intravenous immunoglobulins may normalize serum hyper IgE and increase neutrophil chemotaxis. Monoclonal anti-IgE treatment has been shown to reduce serum IgE levels, although the pros and cons of this approach are still unclear (2). In this current case study, abscess incision and drainage were required in order to reduce pain and inflammation. In addition, although photodynamic therapy (PDT) leads to excellent results in the treatment of perforated folliculitis, its therapeutic potential in cases of hyper IgE syndrome has not been mentioned in any previous research. We are curious to see if PDT will eventually become a recognized treatment for HIES.
Project supported by the National Science Foundation for Young Scientists of China (Grant No.81500187, No.81502710).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize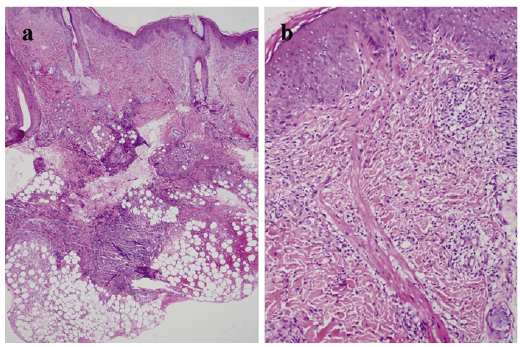 Click to show fullsize
Click to show fullsize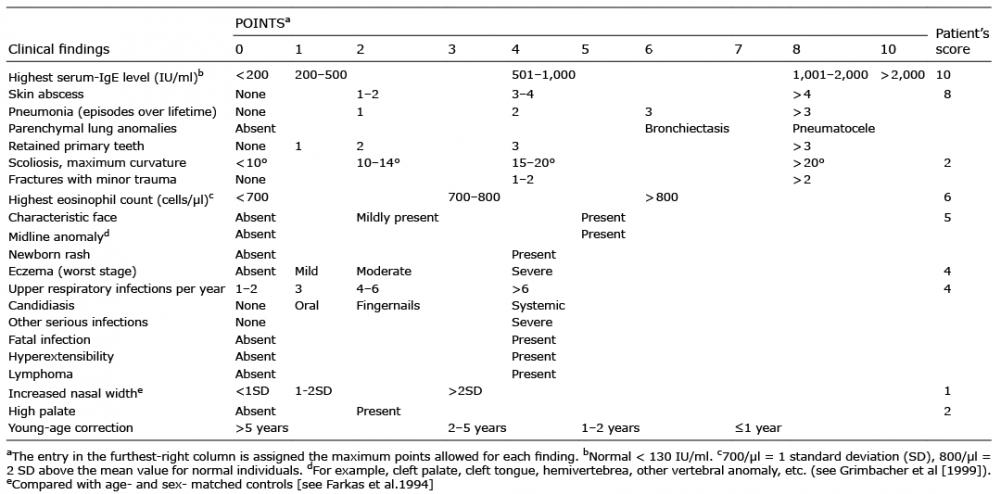 Click to show fullsize
Click to show fullsize