


1Department of Dermatology, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Toyama, Toyama, and 2Department of Dermatology, Hirosaki University Graduate School of Medicine, Hirosaki, Aomori, Japan
Erythropoietic protoporphyria is caused by a partial deficiency of ferrochelatase, which is the last enzyme in the heme biosynthesis pathway. In a typical erythro-poietic protoporphyria, photosensitivity initially appears, following the first exposure to the sun in early infancy or childhood. Erythropoietic protoporphyria has been reported worldwide, but there is a regional variation in its epidemiology. Approximately 20% of the Japanese patients were recognized to have symptoms of erythropoietic protoporphyria after 10 years of age. Physicians occasionally encounter Japanese patients with erythropoietic protoporphyria, mild symptoms and no FECH gene mutations. The homozygous IVS3-48C polymorphism may cause a mild phenotype of the erythropoietic protoporphyria via a slight increase in protoporphyrin. The frequency of the homozygous IVS3-48C polymorphism in the Japanese population is higher than that observed in European countries. Japanese type of erythropoietic protopor-phyria shows a characteristic phenotype of the late onset and mild symptoms compared to the Caucasian erythropoietic protoporphyria. This review describes the characteristics of erythropoietic protoporphyria in Japanese patients.
Key words: erythropoietic protoporphyria; ferrochelatase; photosensitivity.
Accepted Apr 2, 2019; E-published Apr 2, 2019
Acta Derm Venereol
Corr: Megumi Mizawa, M.D., Ph.D., Department of Dermatology, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, Sugitani, Toyama 930-0194, Japan. E-mail: megumiza@med.u-toyama.ac.jp
Erythropoietic protoporphyria (EPP) is a rare autosomal dominant disorder of heme biosynthesis, which results from a decrease in ferrochelatase activity, leading to an excess accumulation of protoporphyrin. EPP is characterized by painful photosensitivity, which begins in early childhood. The time of onset in Japanese patients may be later than in patients of other ethnicities. The homozygous IVS3-48C polymorphism may cause a mild phenotype of EPP via a slight increase in protoporphyrin. Japanese EPP shows a characteristic phenotype, such as the late onset and mild symptoms compared to Caucasian EPP.
Erythropoietic protoporphyria (EPP; OMIM #177000) is an inherited cutaneous porphyria caused by mutations in the ferrochelatase gene (FECH), which codes for an enzyme that catalyses iron insertion into protoporphyrin (PP) to form heme. A partial decrease in FECH activity results in the accumulation of PP in the skin, blood cells and skin blood vessels. The accumulation of PP in the skin causes painful photosensitivity in patients, which begins in early childhood. In less than 10% of patients, the accumulation of PP in the liver can lead to hepatic injury (1, 2). The disease is considered to occur across races and ethnic groups. However, we often note differences between Japanese patients and those from other countries with regard to the clinical symptoms of EPP.
We reviewed the clinical and genetic findings of EPP patients in the Japanese population.
EPP is an autosomal-dominant inherited disorder. More than 175 different mutations of the FECH gene have been reported (3). However, EPP has a low clinical penetrance, with less than 10% of mutation carriers developing overt clinical symptoms (4). Gouya et al. (2, 5, 6) revealed that the single intronic nucleotide polymorphism (SNP) (rs: 2272783) of C at IVS3-48 in trans to a mutated FECH allele results in the development of the EPP phenotype via the low expression of FECH. At present, 12 SNPs in the coding region of FECH gene have been indicated, 4 of which are synonymous (3). It has been shown that the c.68–23C > T variant located in intron 1 (rs: 2269219) alters the secondary structure of the mRNA (7). In addition, the functional signi?cance of c.1-251A>G (rs: 17063905) on the transcriptional activity of the FECH gene lies in decreasing the promoter activity (8).
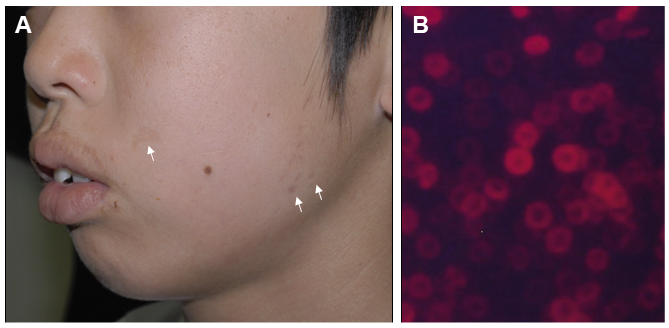
The accumulation of phototoxic PP in the superficial vessels is activated by blue light (400 to 410 nm), triggering singlet oxygen free radical reactions that lead to severe neuropathic pain, which lasts hours to days. PP is released from erythroid cells into the circulation; gains access to the vascular endothelium and liver, and is then excreted through the biliary system. Laboratory investigations have revealed an increased PP concentration. A large number of fluorocytes are observed in the peripheral blood (Fig. 1B). The characteristics of photosensitivity in EPP are ?rst a burning, stinging sensation, appearing immediately at sun exposure, followed by erythema, oedema and petechiae (9–11). Repeated sun exposure can lead to more chronic skin changes, such as a circular or linear scarring and a waxy thickening (Fig. 1A) (9, 11). PP is cleared by the hepatobiliary system and has a concentration-dependent hepatotoxic effect; as such, its accumulation in the liver can impair the hepatobiliary system and potentially lead to liver failure (12). Mild hypochromic microcytic anaemia may also develop (13).
Fig. 1. The clinical finding and fluorocytes of a typical erythropoietic protoporphyria (EPP) patient. A. Small scars on the cheek of an EPP patient (white arrows). B. A large number of fluorocytes in the peripheral blood of a typical EPP patient.
EPP has been reported worldwide; however, there are regional variations in its epidemiology. A minimum prevalence of 1 in 180,000 has recently been reported in Sweden (14) and a slightly higher prevalence has been reported in the European immigrant populations of South Africa (1/152,000) (15), the United Kingdom (1/143,000) (16), Northern Ireland (1/79,000) (17), and the Netherlands (1/75,000) (18). The exact prevalence of EPP in Japan has not been precisely determined yet. However, the penetrance of the EPP phenotype in the Japanese population is expected to be higher than in the Western countries due to the high frequency of the IVS3-48C polymorphism (43%) (19). Conversely, EPP is rare in West Africans because of the extremely low frequency of the IVS3-48C polymorphism (< 1%) (2, 19).
The initial clinical symptoms of EPP usually appear at the first sun exposure in early infancy or childhood. A previous review of 223 EPP cases in the United Kingdom demonstrated the median age of onset to be 1 year (range: 0–12 years) (Table I) (16). In Sweden, the most frequently reported age at the onset of symptoms was the ?rst year of life, and the median age was 3 years (range 0–25 years) (14). North American patients with EPP showed a mean age at the onset of symptoms of 4.1 (standard deviation 3.0) years (n = 158) (20). To examine the age of onset in the Japanese population, we reviewed 103 Japanese EPP cases reported from 1980 to 2015 that were found using Ichushi-Web. Of the 48 papers reviewed, 43 were written in Japanese and 5 in English (21–25). The median age at onset was 6 years (range: 1–25 years; n = 72) (26). Surprisingly, approximately 20% of all patients were recognized as having symptoms of EPP after 10 years of age. We hypothesize that the onset of EPP in the Japanese population is later than that in the Caucasian population. The median ages at the diagnosis were 12 and 22 years in the United Kingdom (16) and Sweden (14), respectively. Although patients are symptomatic in early childhood, there was a delay in the diagnosis, with many patients going undiagnosed or misdiagnosed for several years or even decades. Similarly, the median age at the diagosis was 15 years in Japanese EPP patients (26). Male patients with EPP were reported to have significantly higher PP levels than female patients in the United Kingdom, Sweden and North America (14, 16, 20). However, there was no significant difference in the PP levels between men and women among the Japanese patients (26).
Table I. Clinical features of erythropoietic protoporphyria (EPP) patients in 4 countries
The incidence of cholelithiasis in EPP patients is reported to be as high as 20% and may occur at a young age (4, 27). Conversely, hepatic failure is much less common, with incidence rates anywhere from 2% to 5% (4, 28). In Japanese population studies, 47 patients (51.6%; n = 91) had liver dysfunction, including slight abnormalities in aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and gamma glutamyl transpeptidase (GGT) levels, which were higher than previously reported (14, 16, 20). Twelve patients (13%) had protoporphyric liver failure. Among them, two patients died, and one received a liver transplant. A total of 4 patients (4.4%) had gallstones. Among EPP patients, abnormal liver function test results were reported in 8%, 25% and 26.5% in the United Kingdom (16), Sweden (14) and North America (20), respectively. The percentage of liver dysfunction in Japanese EPP patients was found to be higher than that in other countries. However, the prevalence of liver dysfunction in other countries is the result of a cohort study, while in Japan it is the result of a paper review only. Therefore, comparing those data might have some limitations. Microcytic anaemia is estimated in 20% to 60% of patients (29). Anaemia was reported in 35 Japanese patients with EPP (51.8%; n = 68); it was reported in 21% of Swedish EPP patients (14) and 46.6% of North American EPP patients (20).
There seem to be regional differences in the age at the onset of EPP. The exact mechanism underlying the onset of EPP symptoms remains unclear. Interestingly, few published reports have included adult-onset EPP cases with the FECH gene mutation (30, 31). It was shown that the late-onset EPP patients developed photosensitivity after a strong sun exposure in a tropical climate in middle age, despite not experiencing any symptoms while being in Northern Europe, where they were exposed only to weak sunlight (30, 31). We have also encountered a case in which photosensitivity initially appeared at 13 years of age in a Japanese patient, despite the fact that the patient had fluorocytes, an increased PP level and a FECH gene mutation detected when she was 7 years old (26). In this Japanese patient, we speculate that avoiding sun exposure may have delayed the onset of EPP. Thus, the dose of sun exposure may play an important role in the induction of EPP symptoms.
We hypothesize several possible reasons for the late age of onset in Japanese EPP patients. First, most of the Japanese population tends to avoid strong sun exposure, as recommended by the Japanese Ministry of Environment since 1998. Second, the melanin and carotene content in Japanese skin is greater than in Caucasians skin (32–34). This greater amount of melanin and carotene imbues superior natural photo protection (32, 34, 35). Finally, Japanese EPP patients include a proportion of incomplete EPP patients, who have a mild phenotype and whose symptoms may be less obvious than typical EPP patients as described below.
The FECH gene mutation is detected in approximately 90% of clinically apparent EPP cases. However, in Japan, physicians occasionally encounter EPP patients with a mild photosensitivity associated with a slight increase in the PP concentration, formation of a small number of fluorocytes (Fig. 2A), and no FECH gene mutations. We recently reported 3 Japanese patients with a mild phenotype of EPP. The FECH gene mutation was not detected by direct sequence analyses in any of the patients. However, all patients had the homozygous IVS3-48C polymorphism (36). The homozygous IVS3-48C polymorphism develops a mild phenotype of EPP via a slight increase in PP. We refer to this condition as incomplete EPP. Schneider-Yin et al. (37) also reported an Ashkenazi Jewish patient with the homozygous IVS3-48C polymorphism and no FECH gene mutation, who exhibited both clinical and biochemical signs of a mild phenotype of EPP. Whatley et al. (38) reported a British patient with the homozygous IVS3-48C polymorphism and no FECH gene mutation, who had typical EPP presentation. Furthermore, a recent report showed that 4 of 32 EPP patients had only the homozygous IVS3-48C polymorphism without any other mutations in the FECH gene. They thus showed the classical clinical features (39). In contrast, Colombo et al. (3) described 6 FECH gene mutation non-carriers who were homozygous for the c.1-251G, c.68-23T, and c.315-48C (IVS3-48C) variants and did not show biochemical or clinical symptoms of the disease.
Fig. 2. Fluorocytes and the course of the protoporphyrin (PP) level in incomplete erythropoietic protoporphyria (EPP) patients. A. Only a few fluorocytes were observed in the peripheral blood of an incomplete EPP patient (white arrows). B. The follow-up of course of the PP level in 3 incomplete EPP patients (normal range, 30–86 μg/dl).
The FECH activity was found to be decreased in EPP patients compared to normal subjects, although the values are variable, ranging from 8% to 45% (1, 40). Tahara et al. (41) examined the FECH activity in subjects with no FECH gene mutations. Consequently, the FECH activity in subjects with the IVS3-48C/C and T/C polymorphism were 38% and 50-60% of that observed in patients with the IVS3-48T/T polymorphisms, respectively. Interestingly, Brancaleoni et al. (39) found in their analysis of the percentage of the aberrant inserted FECH mRNA in patients with non-classical EPP (with homozygous IVS3-48C and no FECH gene mutation) that the levels were comparable to those in classical EPP patients (with IVS3-48T/C and the FECH gene mutation). Furthermore, the expression of the FECH gene in non-classical EPP was comparable to that in classical EPP patients (39). Two other variants in the FECH gene (c.1–252A > G and c.68–23C > T) have been found to be associated with the intron 3 variant (7, 8). However, the presence of the G base at position c.1–252 and the T base at position c.68–23 did not seem to in?uence the total expression of the FECH gene (39). Therefore, the homozygous IVS3-48C/C polymorphism was identified as pathological. These findings support the hypothesis that the slight decrease in FECH activity is caused by the IVS3-48C/C polymorphism, resulting in a mild clinical appearance of EPP. Previous studies have speculated that the genotypic frequency of the splice site modulator IVS3-48C may affect the penetrance of the EPP phenotype in affected families as well as the prevalence of the disease in populations.
The frequency of the homozygous IVS3-48C polymor-phism in the Japanese population is over 10 times that of individuals from European countries (19). Therefore, there may be incomplete EPP patients that have not yet been diagnosed in Japan. Furthermore, this may explain the late age of onset in Japanese EPP patients. Indeed, 3 Japanese incomplete EPP patients developed their disease at 5 to 6 years old (36). Of the 103 Japanese EPP patients we mentioned in the previous chapter, 8 had mild photosensitivity and showed a slight increase in their PP levels (<400 μg/dl). Their median age of onset was 3 years (range: 1–15 years), and their median age at the diagnosis was 15 years (range: 1–45 years). How-ever, those patients did not undergo genetic analyses, so the precise details of their condition remain unclear. Nevertheless, we speculate that some or all of them may have had incomplete EPP.
The 3 above-mentioned Japanese incomplete EPP patients were advised to avoid sunlight exposure. They were followed up for their clinical symptoms and PP levels every year for 5 to 8 years (Fig. 2B). There were slight increases in the PP levels over time. Patient 3 occasionally suffered from slight painful photosensitivity in the summer. Nevertheless, in these 3 patients, the frequency and severity of photosensitivity gradually decreased with age, although they were unable to avoid sun exposure completely and sometimes developed mild sunburn. The PP level of patient 2 has decreased almost down to the normal limit. These findings suggest that the symptoms of incomplete EPP patients may improve with age. We need to follow-up the PP level of incomplete EPP carefully in the future. A previous report found that 40% of EPP patients showed an improvement in their symptoms, following a build-up of sunlight tolerance after repeated exposure (42). EPP patients seem to benefit from UV ‘desensitization’ therapy, and anecdotal evidence suggests that natural sunlight exposure improves light tolerance (43). Narrow-band ultraviolet B (UV-B) therapy was reported to increase light protection via epidermal thickening and hyperpigmentation and to suppress the skin’s immune system in EPP patients with a mild form (44). Oral β-carotene at a dose of 60–180 mg/day seems to be effective in clearing singlet oxygen reactive species generated by the photochemical reaction of porphyrin (35) or in protecting the skin from sunlight by skin discoloration due to carotenemia (45). Afamelanotide, a potent α-melanocyte-stimulating hormone (α-MSH) analogue, was approved for treatment of EPP by the European Medicines Agency in 2014. Afamelanotide can darken the skin colour and increase tolerance to sunlight exposure without pain (42, 43). Afamelanotide-induced eumelanin synthesis provided photoprotection and decreased the consequences of phototoxicity (46). These findings support the hypothesis that increased epidermal melanin production and distribution caused by suntan reduces the damaging UV and visible light penetration. Although the exact mechanism underlying the improvement of EPP symptoms remains unclear, we speculate that having a slight suntan can help to improve photosensitivity.
This review demonstrated the clinical features of Japanese EPP patients. Japanese EPP shows a characteristic phenotype, such as late onset and mild symptoms compared to the Caucasian EPP. This may be explained by the existence of incomplete EPP in Japan. To clarify the characteristics of Japanese EPP, further studies are required.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize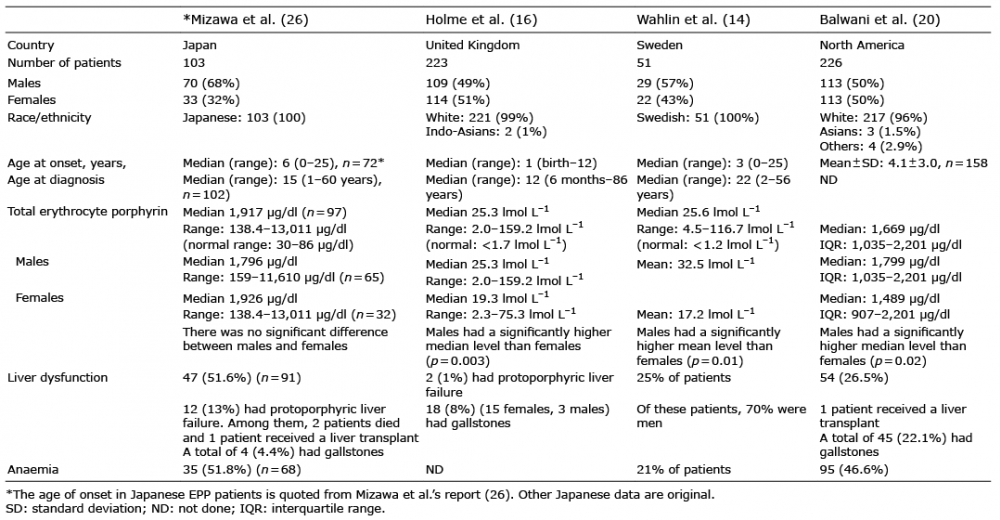 Click to show fullsize
Click to show fullsize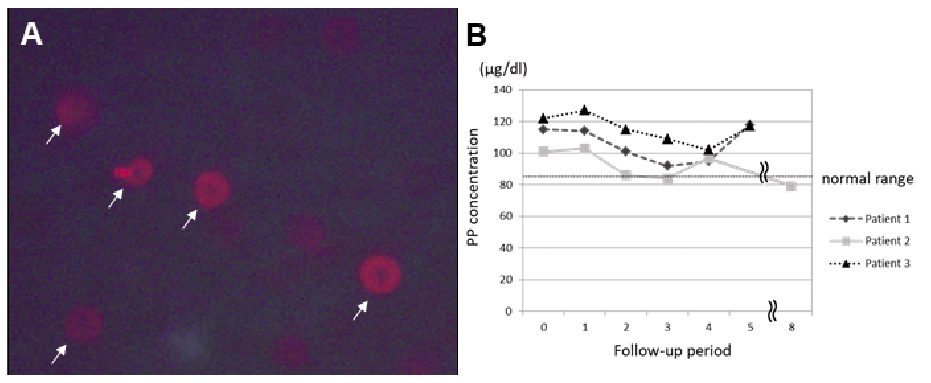 Click to show fullsize
Click to show fullsize